-
大咖云集,成果滿滿 | 2019衡陽精準醫療峰會成功舉行!
2019-06-27 15:25:30
6月25日,由衡陽市非公立醫療協會主辦,深圳百諾精準醫療科技有限公司和衡陽華程醫院共同承辦的“聚力分子生物學 推動醫療新時代”2019衡陽精準醫療峰會暨非公立醫療協會年會,在湖南衡陽神龍大酒店成功舉行。
湖南省民營醫院管理分會原會長張紹金,原國家食藥監管總局處長王鐵,原國家衛生部醫師資格考試辦處長孟磊,張家界市衛生局原局長陳才長,湖南省中醫藥研究院中藥研發部主任尹天雷,懷化市民營醫院協會長石正蘭全權代表鄧源佳,深圳百諾精準醫療科技有限公司CEO余清,深圳精準醫療科技有限公司合作開發事業部總經理鄭堅,深圳精準醫療科技限公司創業合伙人王芳仁院長,衡陽市非公立醫療機構協會會長,華程醫院院長祝平照等醫學界頂級專家以及衡陽地區相關行業協會、醫院的高端醫療部門等相關單位與企業百余位嘉賓共同參與了此次盛會,共同探討精準醫療與精準健康管理面臨的契機與挑戰,把脈精準醫療與智能醫療。

會議在衡陽市非公立醫療機構協會會長祝平照的致辭中拉開帷幕。祝會長總結了近年來,協會的服務成果并表示,我們國家在開展精準醫療與精準醫學方面也有非常好的基礎,但怎樣落實到非公立醫院,今天召開的精準醫療峰會責任很重大,對進一步完善醫院精準醫學研究具有重要的意義。

湖南省民營醫院管理分會原會長、資深民營醫院管理專家張紹金在以《醫療環境與政策》為主題的演講中指出,非公立醫院要持久長遠地發展,必須做好三個工作,一是尊重醫生,關心醫務人員;二是做好基礎工作,加強規章強制度建設;三是構建自己的核心技術體系,只有加強自身建設,才能適應未來醫療環境。

深圳百諾精準醫療科技有限公司CEO余清教授做了題為《攜手核心技術 共拓精準醫療》的精彩報告,對精準醫學與精準健康管理發展提出了自己獨到的見解并分享了百諾精準醫療的使命。余教授表示,精準醫療是醫學自身發展的必然趨勢,精準醫療在腫瘤、全病程精準管理、易感基因風險分析、早篩、分子分型指導精準醫療、腫瘤精準治療等臨床應用已經讓精準醫學引領前沿。面對當下醫療服務四大競爭力準確診斷、有效治療、安全用藥和技術實力,醫院可以通過精準醫學的角度去判斷,把診斷和診療向前推一步。百諾精準醫療“精準檢測、精準解讀、精準應用”三大核心優勢搭載人工智能和大數據技術,能夠賦予醫院在疾病預防、治療、用藥、康復等層面的精準應用,助推醫院在肺癌、肝癌、乳腺癌等重大特定疾病、慢性病方面實現臨床突破。
聽完余教授的精彩演講,已有嘉賓忍不住發問:百諾精準醫療到底是如何為醫院賦能?

對此,深圳百諾精準醫療科技有限公司合作開發事業部總經理鄭堅給出了最完滿的答案。鄭總表示,精準醫療科室未來將是醫院新的增長點,走在技術前沿才有未來!深圳百諾精準醫療科技有限公司是一家專注于精準醫療技術的全球性精準醫療技術服務提供商,百諾精準醫療為醫院搭建“分子醫生”智能化分子醫療技術平臺,孵化精準醫學科室,運用百諾精準檢測、精準解讀、精準應用三大治療與人工智能大數據技術,結合分子學、臨床、人工智能及大數據領域專家多維度的精準解讀,賦予醫院在疾病預防、治療、用藥、康復等層面的精準應用,結合醫院的特殊性,打造屬于醫院自己的精準醫療技術體系,助推醫院國際化精準醫療能力,提升醫院的核心競爭力,造福更多的患者。
隨著鄭堅先生對精準醫療的精彩分享后,百諾精準醫療科技有限公司創業合伙人王芳仁院長與各醫院代表進行了合作簽約,現場響起了雷鳴般的掌聲。

本次峰會共有20余家醫院與百諾精準醫療進行了合作簽約,如衡陽常寧華康醫院,衡陽青山醫院,衡陽華程醫院。未來,百諾精準醫療將在更多城市布局,賦能醫院核心技術,同時,全面推進建立精準醫學科室,推進智慧醫療和社區醫院高質量發展。共同為大健康產業的發展貢獻力量,為人類健康服務!
-
IF=27! Hi-C,發表高分Paper的開掛神器,你需要了解一下!
2018-12-03 11:31:15
Jesse R. Dixon, Jie Xu ,et al. Integrative detection and analysis of structural variation in cancer genomes. Nature Genetics, 2018, 50: 1388–1398. IF 27.125
主要技術:Hi-C、集成光學映射(Irys)、全基因組測序(WGS)
研究背景
結構變異(SVs),包括倒置、刪除、復制陽離子和易位,是大多數癌癥基因組的標志。復發性SVs的發現及其對基因組織和表達的分子效應促進我們對腫瘤發生的認識。許多致癌基因已被確認為復發易位的產物,并為藥物治療特別是造血惡性腫瘤提供了成功的靶點。盡管它很重要,但在癌癥基因組中鑒定SVs仍然具有挑戰性。作者利用高通量染色體構象捕獲(Hi-C)、集成光學映射和全基因組測序,系統地檢測正常或癌癥樣本中的SVs,意圖探究癌基因組中,SVs對突變驅動因素的影響。
研究內容及結果
1. 檢測腫瘤基因組SV的方法
為了評估對SVs不同檢測方法的能力,作者選取了8個癌細胞系和1個典型正常對照(GM12878)(見表1),對它們的WGS、光學測圖和Hi-C數據進行比較(圖1a),發現三種方法均檢測到Caki2細胞中染色體2和3的易位(圖1b),通過觀察同一區域DNA復制時間譜的顯著變化,也證實了這種易位。同時觀察到,與正常細胞相比,癌癥基因組顯示出更多的重組事件,如圖1c所示環狀基因組結構剖面。
表1腫瘤和正常細胞系的高置信度的SVs數

圖1 腫瘤基因組SV檢測的總體策略

2. 利用Hi-C數據檢測大規模重排
在Hi-C實驗中,正常細胞染色體間相互作用非常罕見(圖2a左)。然而,這這種情況在癌細胞中卻相反。例如Caki2癌癥在細胞中,觀察到了強烈的染色體間相互作用(圖2a右),這可能是由于6號染色體和8號染色體的融合。但是關于癌細胞的染色體相互作用增加的信號是由于重組還是三維基因組組織的變異導致的還不清楚,因此針對這一問題,作者首先為“正常”的三維基因組組織特征建立了概率模型,包括位點、TADs、A/B compartments之間的基因組距離,發現小染色體和次端粒區域之間相互作用的增加。并且在重排的情況下,兩個重排區域的基因是融合的,因其改變了位點之間的線性距離,從而也導致了與局部預期交互頻率的偏差(圖2a、b)。其次,作者利用Hi-C數據進行全基因組SVs檢測,這一檢測屬于一種新型算法。該算法具體體現為:作者首先用一個特征良好的慢性粒細胞白血病細胞系(K562)來評估,并將結果與已發表的核型進行比較。在19個Hi-C預測的重排中,11個可以確認,其余8個是新的。由于這8個均在兩個獨立實驗室進行的,它們不太可能是克隆進化的產物。隨后,作者進行了FISH實驗來驗證新的預測易位。使用Hi-C數據預測的19個易位中有18個通過FISH或以前的核型驗證,結果表明,新算法能夠識別具有高特異性的大規模結構變異。
最后,將Hi-C分析擴展到27個癌細胞系和9個核型正常細胞系(圖2d),發現在癌細胞中報告了25次重排,在正常細胞中幾乎沒有發生這種情況,染色體間和染色體內重排的比率約為2:1(所有細胞系中為424比274)。因此,新算法似乎可以識別大部分的大SVs,只有4.3%的無法識別。
圖2 利用Hi-C數據檢測大規模重排


3. 不同方法檢測SVs的比較
通過光學映射和WGS在每個癌細胞株中鑒定了數千個遺傳物質的增加或損耗,光學映射檢測到的缺失比WGS更少但范圍更大。在T47D細胞中,WGS檢測到2943個缺失,中位大小為552 bp,而Irys檢測到1128個缺失,中位大小為1 335 bp(圖3a,b)。其中85% WGS檢測到的缺失被Irys遺漏,且其中78%的中位大小小于1kb。由于其分辨率受兩個刻痕點之間的最小距離的限制,這些特征很可能被光學映射所忽略。Irys預測的缺失中有3%與多個較小的WGS缺失重疊,在這些情況下,這些WGS缺失的總和大小接近Irys檢測到的缺失,但在Irys檢測到的缺失中,有15%沒有被WGS捕獲。
作者測試了Irys檢測到的一部分缺失,其中87.5%的缺失(16個缺失中的14個)通過了PCR驗證。光學映射可以識別WGS reads沒有被映射的重復區域內的缺失(圖3c),以及在斷點周圍可映射性較低的區域。同時還發現WGS、Irys和Hi-C可以檢測到不同染色體間大規模重排,類似基因組的非模板化添加堿基或外源DNA序列,如病毒的堿基,它可能來自第三條染色體因其太短而無法識別。如圖3d所示,光學映射到局部結構,WGS用來確定斷點,WGS通過定位斷點和Hi-C數據來驗證同一等位基因上幾個相鄰重排。總之,采用互補技術的綜合方法對于更全面地了解癌癥基因組的結構變化至關重要。
圖3 不同方法檢測SVs的比較

4. SVs對增強子的影響
科研集市-1 利用CoIP-MS高效篩選體內互作蛋白
2018-07-30 14:42:08
眾所周知,Co-IP是經典的利用抗體從樣品中捕獲靶蛋白及其互作蛋白、復合體的一項技術,能夠特異性富集所研究的目的蛋白。由于過程中采用了非變性條件,保留了互作及復合體的細胞內狀態。相對于酵母雙雜交、pull down等方法,其特色之一便是捕獲的蛋白互作發生在所研究的特定組織細胞內,保存了互作蛋白及復合體的“內源”狀態。本文重在介紹Co-IP同蛋白質譜技術聯用,進行高效篩選鑒定未知互作蛋白。經典Co-IP流程在收集捕獲產物之后,通過SDS-PAGE、銀染分析差異條帶、Western Blot來鑒定某些預設的互作蛋白。這種途徑限于通量、抗體等因素,適合點對點驗證。CoIP-MS即為將免疫共沉淀和蛋白質譜技術串聯,運用LC-MS/MS技術對Co-IP所捕獲的蛋白產物進行鑒定分析的技術。相比于MALDI-TOF, LC-MS/MS方法具有更高的靈敏度和幾乎100%的可靠性,是主流文獻中最常用的方法。在對樣本進行非變性條件的蛋白提取之后,Co-IP所獲產物進行SDS-PAGE分離不同大小的蛋白,蛋白質還原烷基化及酶解,除鹽之后的酶解產物進行LC-MS/MS鑒定,將下機數據進行數據庫檢索,獲取產物中蛋白信息。之后再進一步進行對照組及樣品組的韋恩分析,鑒定蛋白質的GO、COG和KEGG注釋分析,以預測蛋白質的功能,揭示蛋白質在各個生命活動中的生物學意義。 (引自Keller-Pinter A, Ughy B, Domoki M,et al. The phosphomimetic mutation of syndecan-4 binds and inhibits Tiam1 modulating Rac1 activity in PDZ interaction-dependent manner. PLoS One. 2017 Nov 9;12(11):e0187094.)1. 樣品蛋白提取為了保持目的蛋白本身及其互作蛋白、復合體的天然狀態,不遺漏互作蛋白信息,Co-IP樣品的蛋白提取應注意:不使用SDS、Sodium deoxycholate等離子型去垢劑,不能使用尿素、硫脲等強變性劑,也不能經過丙酮/TCA沉淀等導致蛋白變性的有機試劑處理。只能使用如NP-40、Triton X-100等非離子型去垢劑。此外,包括凍融、超聲處理等條件也會不同程度破壞蛋白復合物的存在狀態。因此,如何既保護蛋白活性又獲取較高的提取效率,需要借助經驗及預實驗來確定實驗條件。2. 免疫共沉淀Co-IP在Co-IP過程中,除了所用抗體性能達到IP級別的親和力是先決條件之外,孵育體系中的總蛋白濃度、抗體用量、漂洗條件等方面也決定Co-IP過程的靈敏度以及特異性。通常總蛋白濃度需要≥1mg/ml,當總蛋白濃度足夠高時,可以適當倍數稀釋樣品以降低其中去垢劑濃度,為抗體反應提供溫和環境。使用protein A/G的磁珠或凝膠時不宜貪多,目前商品化產品其載量都在50ul用量時承載50ug或更多抗體,而通常一個Co-IP反應的用量在1-3ug左右。漂洗緩沖液需要控制適當的鹽離子強度、去垢劑種類及濃度,通常會延用裂解液緩沖液配方,因為背景蛋白在后續質譜過程中會干擾蛋白的鑒定,優化漂洗緩沖液的目的是既保持目的蛋白復合物的捕獲效率又盡量減少非特異結合蛋白。3. 質譜上機前樣本處理蛋白質還原烷基化和酶解。蛋白質的還原烷基化如下,加入二硫蘇糖醇(DTT)還原蛋白質,再加入碘乙酸銨(IAM),最后加入Trypsin酶,過夜酶解,酶解后處理。酶解產生的多肽用C18柱子除鹽,已經除鹽的多肽抽干后用Loading Buffer 溶解多肽。多肽上LC-MS/MS儀器進行分析,然后對結果進行評估。
(引自Keller-Pinter A, Ughy B, Domoki M,et al. The phosphomimetic mutation of syndecan-4 binds and inhibits Tiam1 modulating Rac1 activity in PDZ interaction-dependent manner. PLoS One. 2017 Nov 9;12(11):e0187094.)1. 樣品蛋白提取為了保持目的蛋白本身及其互作蛋白、復合體的天然狀態,不遺漏互作蛋白信息,Co-IP樣品的蛋白提取應注意:不使用SDS、Sodium deoxycholate等離子型去垢劑,不能使用尿素、硫脲等強變性劑,也不能經過丙酮/TCA沉淀等導致蛋白變性的有機試劑處理。只能使用如NP-40、Triton X-100等非離子型去垢劑。此外,包括凍融、超聲處理等條件也會不同程度破壞蛋白復合物的存在狀態。因此,如何既保護蛋白活性又獲取較高的提取效率,需要借助經驗及預實驗來確定實驗條件。2. 免疫共沉淀Co-IP在Co-IP過程中,除了所用抗體性能達到IP級別的親和力是先決條件之外,孵育體系中的總蛋白濃度、抗體用量、漂洗條件等方面也決定Co-IP過程的靈敏度以及特異性。通常總蛋白濃度需要≥1mg/ml,當總蛋白濃度足夠高時,可以適當倍數稀釋樣品以降低其中去垢劑濃度,為抗體反應提供溫和環境。使用protein A/G的磁珠或凝膠時不宜貪多,目前商品化產品其載量都在50ul用量時承載50ug或更多抗體,而通常一個Co-IP反應的用量在1-3ug左右。漂洗緩沖液需要控制適當的鹽離子強度、去垢劑種類及濃度,通常會延用裂解液緩沖液配方,因為背景蛋白在后續質譜過程中會干擾蛋白的鑒定,優化漂洗緩沖液的目的是既保持目的蛋白復合物的捕獲效率又盡量減少非特異結合蛋白。3. 質譜上機前樣本處理蛋白質還原烷基化和酶解。蛋白質的還原烷基化如下,加入二硫蘇糖醇(DTT)還原蛋白質,再加入碘乙酸銨(IAM),最后加入Trypsin酶,過夜酶解,酶解后處理。酶解產生的多肽用C18柱子除鹽,已經除鹽的多肽抽干后用Loading Buffer 溶解多肽。多肽上LC-MS/MS儀器進行分析,然后對結果進行評估。 (引自Chen R, Xiao M, Gao H, et al. Identification of a novel mitochondrial interacting protein of C1QBP using subcellular fractionation coupled with CoIP-MS.[J]. Analytical & Bioanalytical Chemistry, 2016, 408(6):1557-1564.)4. 數據檢索、蛋白鑒定LC-MS/MS下機后,將原始下載數據直接提交到與質譜儀連接的Proteinpilot軟件中進行數據庫檢索,過濾掉常見污染蛋白及與之匹配的肽段,得到每個樣品鑒定到的肽段和蛋白結果。通常CoIP-MS是將IgG組及IP同時進行質譜鑒定,得到的反應兩組蛋白差異的韋恩圖。5. 鑒定蛋白功能注釋對鑒定到的蛋白質進行GO、COG和KEGG注釋分析,以預測蛋白質的功能,揭示蛋白質在各個生命活動中的生物學意義。其中,GO分析總共有三個本體(ontology),分別描述基因的分子功能(molecular function)、細胞組分(cellular component)、參與的生物過程(biological process);COG是對蛋白質進行直系同源分類的數據庫;KEGG是有關Pathway的主要公共數據庫,通過Pathway分析能確定蛋白質參與的最主要生化代謝途徑和信號轉導途徑。
(引自Chen R, Xiao M, Gao H, et al. Identification of a novel mitochondrial interacting protein of C1QBP using subcellular fractionation coupled with CoIP-MS.[J]. Analytical & Bioanalytical Chemistry, 2016, 408(6):1557-1564.)4. 數據檢索、蛋白鑒定LC-MS/MS下機后,將原始下載數據直接提交到與質譜儀連接的Proteinpilot軟件中進行數據庫檢索,過濾掉常見污染蛋白及與之匹配的肽段,得到每個樣品鑒定到的肽段和蛋白結果。通常CoIP-MS是將IgG組及IP同時進行質譜鑒定,得到的反應兩組蛋白差異的韋恩圖。5. 鑒定蛋白功能注釋對鑒定到的蛋白質進行GO、COG和KEGG注釋分析,以預測蛋白質的功能,揭示蛋白質在各個生命活動中的生物學意義。其中,GO分析總共有三個本體(ontology),分別描述基因的分子功能(molecular function)、細胞組分(cellular component)、參與的生物過程(biological process);COG是對蛋白質進行直系同源分類的數據庫;KEGG是有關Pathway的主要公共數據庫,通過Pathway分析能確定蛋白質參與的最主要生化代謝途徑和信號轉導途徑。天然和人工培育冬蟲夏草的比較蛋白質組學表征及其營養價值評估
2018-07-30 14:17:12
題目:A comparative proteomic characterization and nutritional assessment of naturally-and artificially-cultivated Cordyceps sinensis.Journal of Proteomics. IF 3.722 Label-free研究背景冬蟲夏草因其營養和藥用價值而受到越來越多的關注。近期,有報道稱,成功通過人工方式培育出冬蟲夏草。本文主要使用非標定量Label-free蛋白質組學技術研究自然生長和人工培育的冬蟲夏草之間的蛋白質組學差異,揭示冬蟲夏草蛋白質組學特征,了解其發育感染過程,并為人工培育提供參考信息。研究內容和結果1. 蛋白組學數據結果及蛋白質控人工培育的蟲草和青藏高原上自然生長的蟲草在形態特征高度相似(圖1),并各自選擇3個批次的樣本進行蛋白提取。將蟲草分為蟲體和子座,分別用SDS-PAGE對樣本蛋白進行質控檢測(圖2)。對解析的蛋白質組學數據進行質控分析,肽段質量容差為0.1Da,說明質譜鑒定肽段準確度高,肽段長度范圍為8-38個氨基酸,分子量范圍為400-12000Da,與預設分子量范圍一致。在本次實驗中,大多數鑒定蛋白含有2-20個unique肽段。
 使用變異系數(CV)驗證實驗方法的重復性,在本次實驗結果中,人工培育的蟲體(AC)、子座(AS)、天然生長的蟲體(NC)、子座(NS)的CV值中位數分別為31.88%,27.44%,34.49%,34.94%。12個樣品的相關性系數在0.68-0.93之間,這些結果證明了實驗的可重復性。在所有的鑒定蛋白中,基于強度的絕對定量(iBAQ)值分布在1200中最豐富的蛋白質中(圖3A)。所有樣品和組內蛋白質的平均iBAQ值相似(圖3B)。這些結果表明蛋白質組內比較可能不受蛋白質豐度系統差異的影響。
使用變異系數(CV)驗證實驗方法的重復性,在本次實驗結果中,人工培育的蟲體(AC)、子座(AS)、天然生長的蟲體(NC)、子座(NS)的CV值中位數分別為31.88%,27.44%,34.49%,34.94%。12個樣品的相關性系數在0.68-0.93之間,這些結果證明了實驗的可重復性。在所有的鑒定蛋白中,基于強度的絕對定量(iBAQ)值分布在1200中最豐富的蛋白質中(圖3A)。所有樣品和組內蛋白質的平均iBAQ值相似(圖3B)。這些結果表明蛋白質組內比較可能不受蛋白質豐度系統差異的影響。 2. 天然蟲草和人工培育蟲草的蛋白表達圖譜在本次實驗中,12個樣品共鑒定到22829條可信肽段(conf ≥ 95%),2541個蛋白質。使用PCA分析天然蟲草和人工培育蟲草蛋白表達圖譜特征,發現雖然蟲體和子座蛋白質分布有明顯區別,但是天然蟲草和人工培育蟲草的整體蛋白質分布類似(圖4)。COG功能注釋到了1951個蛋白質。這些蛋白質主要參與能量產生、轉化,氨基酸轉運、代謝和轉錄調節(圖5)。
2. 天然蟲草和人工培育蟲草的蛋白表達圖譜在本次實驗中,12個樣品共鑒定到22829條可信肽段(conf ≥ 95%),2541個蛋白質。使用PCA分析天然蟲草和人工培育蟲草蛋白表達圖譜特征,發現雖然蟲體和子座蛋白質分布有明顯區別,但是天然蟲草和人工培育蟲草的整體蛋白質分布類似(圖4)。COG功能注釋到了1951個蛋白質。這些蛋白質主要參與能量產生、轉化,氨基酸轉運、代謝和轉錄調節(圖5)。
 圖5 COG功能注釋結果3. 顯著差異蛋白質鑒定結果本次實驗中,顯著差異蛋白數目分別為107(NC/AC)、58(NS/AS),195(AC/AS)、168(NC/NS)。使用層次聚類分析蛋白質表達變化趨勢,主要聚成三類,Cluster I 由在AS中增加并在NC中減少的蛋白質組成;Cluster II包括在AC中降低并在NS中增加的蛋白質;Cluster III包括在AC或NC中增加并在AS和NS中均降低的蛋白質(圖6)。為了確定這些蛋白質的功能特性,將它們的序列映射到GO數據庫。GO富集表明這些差異豐富的蛋白質主要參與生物調節、單一生物過程,結合、催化活性,并定位于細胞器、細胞膜。結果說明這些顯著差異蛋白在蟲草生命活動中主要發揮催化和結合作用(圖7)。
圖5 COG功能注釋結果3. 顯著差異蛋白質鑒定結果本次實驗中,顯著差異蛋白數目分別為107(NC/AC)、58(NS/AS),195(AC/AS)、168(NC/NS)。使用層次聚類分析蛋白質表達變化趨勢,主要聚成三類,Cluster I 由在AS中增加并在NC中減少的蛋白質組成;Cluster II包括在AC中降低并在NS中增加的蛋白質;Cluster III包括在AC或NC中增加并在AS和NS中均降低的蛋白質(圖6)。為了確定這些蛋白質的功能特性,將它們的序列映射到GO數據庫。GO富集表明這些差異豐富的蛋白質主要參與生物調節、單一生物過程,結合、催化活性,并定位于細胞器、細胞膜。結果說明這些顯著差異蛋白在蟲草生命活動中主要發揮催化和結合作用(圖7)。
 4. 顯著差異蛋白Pathway Enrichment 結果為了進一步研探索發生變化的生物學途徑,使用KEGG Pathway 數據庫進行富集分析,結果表明,天然蟲草和人工培育蟲草之間的差異蛋白主要參與次生代謝物合成、丙酮酸代謝、賴氨酸生物合成。在氨基酸合成、代謝途徑中(圖8),有7種相關蛋白發生表達量變化,其中,核糖5-磷酸異構酶(K01807)和丙酮酸激酶(K00873)在NC中降低,而NADH(P)結合蛋白(K00831)在NC中增加; NS中糖基轉移酶(K00766)和雄性不育蛋白(K00143)增加;異檸檬酸/異丙基蘋果酸脫氫酶(K00052)在NC中降低但在NS中升高,精氨酸琥珀酸合成酶(K01940)在NC和NS中均下降。
4. 顯著差異蛋白Pathway Enrichment 結果為了進一步研探索發生變化的生物學途徑,使用KEGG Pathway 數據庫進行富集分析,結果表明,天然蟲草和人工培育蟲草之間的差異蛋白主要參與次生代謝物合成、丙酮酸代謝、賴氨酸生物合成。在氨基酸合成、代謝途徑中(圖8),有7種相關蛋白發生表達量變化,其中,核糖5-磷酸異構酶(K01807)和丙酮酸激酶(K00873)在NC中降低,而NADH(P)結合蛋白(K00831)在NC中增加; NS中糖基轉移酶(K00766)和雄性不育蛋白(K00143)增加;異檸檬酸/異丙基蘋果酸脫氫酶(K00052)在NC中降低但在NS中升高,精氨酸琥珀酸合成酶(K01940)在NC和NS中均下降。 為了驗證組學結果,LC-MS分析蟲草代謝產物。根據代謝組學結果,共鑒定出8種氨基酸,其中4種氨基酸含量發生顯著改變,具體為賴氨酸,精氨酸,蘇氨酸和絲氨酸(圖9)。氨基酸的變化趨勢跟上游合成它們的酶的變化趨勢是一直的。根據中國藥典中關于測定核苷和核苷酸含量,特別是腺苷的規定,NC/NS以及AC/AS總離子色譜圖中沒有觀察到顯著差異(圖10)。
為了驗證組學結果,LC-MS分析蟲草代謝產物。根據代謝組學結果,共鑒定出8種氨基酸,其中4種氨基酸含量發生顯著改變,具體為賴氨酸,精氨酸,蘇氨酸和絲氨酸(圖9)。氨基酸的變化趨勢跟上游合成它們的酶的變化趨勢是一直的。根據中國藥典中關于測定核苷和核苷酸含量,特別是腺苷的規定,NC/NS以及AC/AS總離子色譜圖中沒有觀察到顯著差異(圖10)。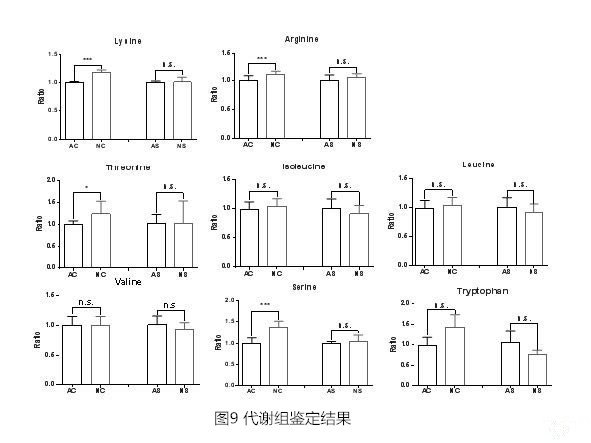
 蟲草蛋白質中必須氨基酸比例與人類蛋白質中的比例相似,說明它具有營養價值。而且,天然蟲草和人工培育蟲草蛋白質中氨基酸成分和含量幾乎相同,沒有顯著差異。文章小結該研究提供了天然蟲草和人工培育蟲草的蛋白表達圖譜,發現天然蟲草和人工培育蟲草的真菌中的蛋白質和代謝產物組成相似,而蟲體和子座的蛋白質和代謝產物方面則有區別。蛋白質組學結果表明氨基酸合成和代謝途徑受到栽培方式的影響。 這些蛋白質組學數據將有助于理解冬蟲夏草的藥用價值,并為其人工培育提供參考。參考文獻1. Shrestha UB, Bawa KS. Trade, harvest, and conservation of caterpillar fungus (Ophiocordyceps sinensis) in the Himalayas. Biol Conserv 2013;159:514.2. Zhao J, Xie J, Wang LY, Li SP. Advanced development in chemical analysis of Cordyceps. J Pharm Biomed Anal 2014;87:271–89.3. Hu HK, Xiao L, et al. Identification of chemical markers in Cordyceps sinensis by HPLC-MS/MS. Anal and Bioanal Chem 2015;407:8059–8066.4. Bereman MS, Beri J, Sharma V. An automated pipeline to monitor system performance in liquid chromatography-tandem mass spectrometry proteomic experiments. J Proteome Res 2016;15:4763-9.5. Xia E-H, Yang D-R, et al. The caterpillar fungus, Ophiocordyceps sinensis, genome provides insights into highland adaptation of fungal pathogenicity. Sci Rep 2017;7:1806.
蟲草蛋白質中必須氨基酸比例與人類蛋白質中的比例相似,說明它具有營養價值。而且,天然蟲草和人工培育蟲草蛋白質中氨基酸成分和含量幾乎相同,沒有顯著差異。文章小結該研究提供了天然蟲草和人工培育蟲草的蛋白表達圖譜,發現天然蟲草和人工培育蟲草的真菌中的蛋白質和代謝產物組成相似,而蟲體和子座的蛋白質和代謝產物方面則有區別。蛋白質組學結果表明氨基酸合成和代謝途徑受到栽培方式的影響。 這些蛋白質組學數據將有助于理解冬蟲夏草的藥用價值,并為其人工培育提供參考。參考文獻1. Shrestha UB, Bawa KS. Trade, harvest, and conservation of caterpillar fungus (Ophiocordyceps sinensis) in the Himalayas. Biol Conserv 2013;159:514.2. Zhao J, Xie J, Wang LY, Li SP. Advanced development in chemical analysis of Cordyceps. J Pharm Biomed Anal 2014;87:271–89.3. Hu HK, Xiao L, et al. Identification of chemical markers in Cordyceps sinensis by HPLC-MS/MS. Anal and Bioanal Chem 2015;407:8059–8066.4. Bereman MS, Beri J, Sharma V. An automated pipeline to monitor system performance in liquid chromatography-tandem mass spectrometry proteomic experiments. J Proteome Res 2016;15:4763-9.5. Xia E-H, Yang D-R, et al. The caterpillar fungus, Ophiocordyceps sinensis, genome provides insights into highland adaptation of fungal pathogenicity. Sci Rep 2017;7:1806.iTRAQ定量蛋白質組學:分析SEMA3B-AS1抑制人骨髓間充質干細胞成骨分化機制
2018-07-30 13:57:05
題目:SEMA3B-AS1-inhibited osteogenic differentiation of human mesenchymal stem cells revealed by quantitative proteomics analysis.Journal of cellular physiology. IF 3.923 iTRAQ研究背景常見的非編碼RNA包括lncRNA、circRNA、miRNA,在細胞的生長、增殖、分化、凋亡中具有重要的調控作用。研究發現,非編碼RNA與腫瘤的發生、發展、轉移密切相關。作為非編碼RNA的一員,lncRNA已被證實廣泛在各類腫瘤的發生發展中扮演重要角色。新骨形成被認為是治療骨相關疾病的一種新型的可替代方法,例如骨質疏松、骨架變形、骨折等。許多研究旨在提高新骨形成過程以希望治愈這些疾病。骨髓間充質干細胞(hMSC)是具有分化為成骨細胞、軟骨細胞展現多種分化潛能的成纖維樣多能干細胞,在新骨形成過程和骨質疏松發病等過程中發揮重要的作用。長鏈非編碼RNA(lncRNAs)被發現參與成骨分化及骨質疏松的過程,比如H19通過吸附miRNA-675促進成骨細胞分化,MEG3通過靶向BMP4或者miR-140-5p來提高MSC的骨生成。但是lncRNA參與hMSC分化過程以及具體的機制目前不是很清楚。hMSC的分化是接受不同環境刺激后發生的過程,細胞表面受體在這個過程中發揮重要的作用,可以起到信號級聯及放大的作用。SEMA家族蛋白被發現在骨生成或者骨疾病中發揮作用。如Sema6d、mema4d、sema3a等被發現可以平衡破骨細胞和成骨細胞來調節骨形成和骨吸收。前期研究發現SEMA3B-AS1可以抑制hMSC增殖和骨分化,具體機制仍需研究。目前lncRNA研究手段主要有:① 功能、表型、疾病性相關研究;②iTRAQ蛋白質組學分析lncRNA差異表達、預測新的lncRNA、生信分析lncRNA靶基因;③ 互作研究及質譜尋找及驗證與lncRNA結合的RNA及蛋白等。腫瘤等疾病中lncRNA作用機制研究有望為疾病的治療尋找新的靶點。本研究主要采用iTRAQ蛋白組學研究反義lncRNA-SEMA3B-AS1的作用機制。研究內容及結果1. MTT法檢測細胞增殖和茜素紅S染色實驗證實SEMA3B-AS1可以抑制細胞的增殖(a)和向成骨轉化(b)。說明SEMA3B-AS1具有抑制人骨髓間充質干細胞增殖和向成骨分化的功能作用。 2. 上述功能驗證實驗已經確定了SEMA3B-AS1對人骨髓干細胞的增殖和分化產生了影響。但是SEMA3B-AS1通過何種途徑發揮功能呢?作者接著包裝SEMA3B-AS1過表達慢病毒,采用iTRAQ蛋白組學技術檢測過表達SEMA3B-AS1和空載體的兩個細胞組中差異蛋白變化。通過對差異蛋白做了聚類和功能富集,發現SEMA3B-AS1可能通過上調或下調相關基因來影響肌動蛋白骨架、黏著斑、胞外基質與受體的相互作用,來調節細胞的骨生成,進而影響細胞增殖和分化。
2. 上述功能驗證實驗已經確定了SEMA3B-AS1對人骨髓干細胞的增殖和分化產生了影響。但是SEMA3B-AS1通過何種途徑發揮功能呢?作者接著包裝SEMA3B-AS1過表達慢病毒,采用iTRAQ蛋白組學技術檢測過表達SEMA3B-AS1和空載體的兩個細胞組中差異蛋白變化。通過對差異蛋白做了聚類和功能富集,發現SEMA3B-AS1可能通過上調或下調相關基因來影響肌動蛋白骨架、黏著斑、胞外基質與受體的相互作用,來調節細胞的骨生成,進而影響細胞增殖和分化。 圖3. 功能富集和差異蛋白表達分析
圖3. 功能富集和差異蛋白表達分析 圖3. 功能富集和差異蛋白表達分析文章小結lncRNA分為正義lncRNA和反義lncRNA,同正義lncRNA相比,反義lncRNA由于與正義鏈互補,反義鏈lncRNA與相鄰或相近基因的調控關系更加密切。首先,作者以骨質疏松發病機制研究為目的,以人骨髓間充質干細胞、反義lncRNA(SEMA3B-AS1)為對象,進行功能實驗,檢測了SEMA3B-AS1對細胞增殖和骨生成的影響。同時,作者采用iTRAQ蛋白組學技術檢測分析受SEMA3B-AS1調控的蛋白差異表達及差異蛋白的生物學過程及相關的代謝通路。最后,作者分析了SEMA3B-AS1調節的骨生成在骨質疏松發生中的作用,得出SEMA3B-AS1可能是骨質疏松治療的一個靶點。參考文獻1. J.C. Chen, C.R. Jacobs, Mechanically induced osteogenic lineage commitment of stem cells, Stem Cell Res Ther, 4 (2013) 107.2. J.J. Quinn, H.Y. Chang, Unique features of long non-coding RNA biogenesis and function, Nat Rev Genet, 17 (2016) 47-62.3. C. Sang, Y. Zhang, F. et al , Tumor necrosis factor alpha suppresses osteogenic differentiation of MSCs by inhibiting semaphorin 3B via Wnt/beta-catenin signaling in estrogen-deficiency induced osteoporosis, Bone, 84 (2016) 78-87.4. P.S. Mathieu, E.G. Loboa, Cytoskeletal and focal adhesion influences on mesenchymal stem cell shape, mechanical properties, and differentiation down osteogenic, adipogenic, and chondrogenic pathways, Tissue Eng Part B Rev, 18 (2012) 436-444.
圖3. 功能富集和差異蛋白表達分析文章小結lncRNA分為正義lncRNA和反義lncRNA,同正義lncRNA相比,反義lncRNA由于與正義鏈互補,反義鏈lncRNA與相鄰或相近基因的調控關系更加密切。首先,作者以骨質疏松發病機制研究為目的,以人骨髓間充質干細胞、反義lncRNA(SEMA3B-AS1)為對象,進行功能實驗,檢測了SEMA3B-AS1對細胞增殖和骨生成的影響。同時,作者采用iTRAQ蛋白組學技術檢測分析受SEMA3B-AS1調控的蛋白差異表達及差異蛋白的生物學過程及相關的代謝通路。最后,作者分析了SEMA3B-AS1調節的骨生成在骨質疏松發生中的作用,得出SEMA3B-AS1可能是骨質疏松治療的一個靶點。參考文獻1. J.C. Chen, C.R. Jacobs, Mechanically induced osteogenic lineage commitment of stem cells, Stem Cell Res Ther, 4 (2013) 107.2. J.J. Quinn, H.Y. Chang, Unique features of long non-coding RNA biogenesis and function, Nat Rev Genet, 17 (2016) 47-62.3. C. Sang, Y. Zhang, F. et al , Tumor necrosis factor alpha suppresses osteogenic differentiation of MSCs by inhibiting semaphorin 3B via Wnt/beta-catenin signaling in estrogen-deficiency induced osteoporosis, Bone, 84 (2016) 78-87.4. P.S. Mathieu, E.G. Loboa, Cytoskeletal and focal adhesion influences on mesenchymal stem cell shape, mechanical properties, and differentiation down osteogenic, adipogenic, and chondrogenic pathways, Tissue Eng Part B Rev, 18 (2012) 436-444.蛋白質組學研究:揭示人調節T細胞在保護細胞信號通路中的適應性
2018-07-30 13:35:59
Proteomic Analyses of Human Regulatory T Cells Reveal Adaptations in Signaling Pathways that Protect Cellular Identity.Immunity IF19.734 蛋白質組研究背景
Treg cells(調節性T細胞)因其轉錄因子FOXP3的表達和抑制免疫應答的能力構成了獨特的CD4+T細胞譜系。Treg 細胞可以保護免疫耐受,抑制免疫細胞造成的組織損傷,促進組織修復。Treg細胞也有不好的一面,它可以阻礙機體對癌細胞的免疫能力,有研究認為Treg這部分的功能缺失可能是造成人類自身免疫疾病和過敏發生的基礎。因此,需要有針對性的促進或抑制人類疾病中的Treg細胞功能。Treg細胞和Tconv細胞密切相關,作者希望能夠在分子水平上對二者進行詳細研究和區分。研究內容及結果
1. Treg細胞蛋白表達特征
作者從健康人供體的外周血單核細胞(PBMCs)分離CD4 + T細胞亞群。這些細胞亞群包含nTconv細胞(CD45RA +CD25-)、記憶(m)Tconv細胞(CD45RA-CD25-)、nTreg細胞(CD45RA+CD25hi)和eTreg細胞(CD45RA-CD25hi)(圖1A)。兩種Treg細胞亞群都表達FOXP3和Helios并且缺乏IL-7受體α鏈(CD127),而nTconv和mTconv細胞亞群則相反,它們缺乏FOXP3和Helios,但是能表達CD127。使用高分辨率質譜(MS),作者鑒定了平均35,744±3,757個肽段,5,955±344個蛋白gurup。在所有五個CD4+T細胞亞群中定量了4,358種不同的蛋白質。其中,422種蛋白質基于其非標記定量(LFQ)值在CD4+T細胞亞群中表現出差異表達(FDR <0.05)。 總數據集的主成分分析(PCA,圖1B)、Pearson相關分析(圖1C)、差異表達蛋白的層次聚類(圖1D)證實了生物重復的密切相關性。作者分離CD4+T細胞亞群的原則并不是基于細胞譜系,而是根據CD45RA的表達(圖1D)。富集分析顯示,幼稚Treg和Tconv細胞共享參與磷酸戊糖代謝、NADP代謝和染色質組織等過程中的過表達蛋白(圖1D),而eTreg和mTconv細胞共享參與蛋白質合成、細胞運輸、信號傳導和凋亡的過表達蛋白(圖1D)。基于分化階段蛋白質表達的相似性可部分反映淋巴組織(幼稚細胞)與非淋巴組織(效應細胞)的優先定位。從聚類結果可以發現具體的Treg細胞蛋白特征,如nTreg和eTreg細胞共享某些高表達蛋白(cluster1)和低表達蛋白(cluster9-10),而eTreg細胞則獨自表現出在cluster2中蛋白高表達以及在cluster6中蛋白低表達。 圖1 CD4+T細胞亞群蛋白質組學結果2. Treg細胞的mRNA表達特征
圖1 CD4+T細胞亞群蛋白質組學結果2. Treg細胞的mRNA表達特征
Treg細胞的蛋白組學結果與已發表的相似的細胞亞群的轉錄組學結果關聯性較低。為了進行直接比較,作者對五個CD4+T細胞亞群進行了全基因組mRNA深度測序。也基于它們的轉錄組分析發現,nTreg和nTconv細胞聚集在一起并遠離三種效應和記憶表型CD4+T細胞群(圖2A)。在五個CD4 + T細胞亞群(圖2B)之間總共649個差異表達mRNA(p <0.05),包括預期的標志基因如IL-7R、IKZF2(HELIOS)和效應細胞因子。作者分析發現,eTreg細胞的mRNA表達特征既有特異性(圖2B,cluster7;),也有和其他Treg細胞的一致的共性,如都可以表達FOXP3、IL2RA、TIGIT等分子(圖2B和2C,cluster6)。已經發表的文章結果也證明了作者的發現。作者發現,Treg細胞蛋白質組學結果和轉錄組結果中只有三個分子(FOXP3,SHMT2和SWAP70)表達量趨勢上發生重疊(圖2F)。事實上,553 個mRNA和409個蛋白質在五個CD4+ T細胞亞群之間顯著差異表達,并且可以在兩個水平上定量,但是二者僅重疊48個(圖2G)。因此,與眾多報道的文獻結果一致,相對于轉錄組結果,蛋白質組學會獲得明顯不同的結果。 圖2 CD4+ T細胞亞群mRNA表達結果3. 蛋白質水平與mRNa水平的區別
圖2 CD4+ T細胞亞群mRNA表達結果3. 蛋白質水平與mRNa水平的區別
為了定量比較蛋白質和mRNA水平,作者使用基于強度的絕對定量分析蛋白質數據(iBAQ)。發現蛋白質和mRNA的豐度水平都超過五個數量級(圖3A和3D)。在蛋白水平中,豐度最高的是核糖體和代謝蛋白(圖3B),在mRNA水平中,豐度最高的是編碼參與(免疫)細胞、信號傳導和功能的核糖體組分和分子(圖3E),差異蛋白的豐度通常要比mRNA要高。
作者對4792個mRNA-protein對的表達量在轉錄組和蛋白組水平上進行定位,發現其相關系數約為0.44±0.01。422種差異表達的蛋白質中,在mRNA水平檢測到409種,并進行定量比較(圖3G)。雖然蛋白質和mRNA表達水平通常相關(例如,FOXP3I、KZF2),但是大多數僅有一種(蛋白或mRNA)的表達量達到差異統計標準,這就解釋了差異表達的蛋白質和mRNA之間的有限重疊(圖2F)。作者發現一些分子僅在mRNA或蛋白質水平上真正差異表達(圖3H),這說明細胞調節具有一定的層次范圍。例如,對于FTH1(圖3G),是一個已知在嚴格的翻譯控制下的蛋白質(Hentze等人,2010),它在nTreg和eTreg細胞中豐度都很高;STAM2在mTconv細胞中雖然mRNA水平低,但是蛋白豐度很高(圖3G), 另一方面,在nTreg和eTreg細胞中,mRNA上高表達但蛋白質水平并不高(圖3G)。作者認為聯合蛋白質組和轉錄組分析可以增加對通路分析的可信度。這樣的組合數據有力地論證了與其他CD4+ T細胞亞群相比,核因子kB(NF-kB)和JAK-STAT途徑在eTreg細胞中更容易脫敏(圖3I)。總之,其研究結果強調了蛋白質組學分析對細胞類型功能表征的重要性。 圖3 mRNA和蛋白表達的通路富集分析結果4. Treg細胞具有穩定的蛋白質特征
圖3 mRNA和蛋白表達的通路富集分析結果4. Treg細胞具有穩定的蛋白質特征
作者定義的常見Treg細胞特征由22個具有較高表達的蛋白質和29個低表達蛋白質組成(圖4A)。 免疫印跡和流式細胞術證實了這些蛋白質中的8種蛋白質組學數據。 在早期的Treg細胞蛋白質組學研究中,多是基于CD4+CD127-CD25+和CD4+CD127+ CD25-細胞,覆蓋的蛋白質組較少,所以并沒有找到類似作者發現的組學特征。常見的Treg細胞特征包括FOXP3、IKZF2(Helios)、代謝蛋白GK、UGP2和SHMT2、鐵儲存蛋白鐵蛋白重鏈和輕鏈(FTH1,FLT),以及溶酶體蛋白ASAH1、GGH、GUSB、SGSH和PLBD2,與Tconv細胞相比,它們都在Treg中以高豐度表達(圖4A和4B)。 該特征還包括糖酵解酶HK1、ME2、脂肪酸氧化酶AP00和線粒體脂肪酸轉運蛋白(CPT1A),與Tconv細胞相比,均在Treg中以低豐度表達。作者發現許多信號分子在常見的Treg細胞特征中是差異表達的,如脫氫泛素酶OTULIN的高表達,TNFα誘導的NF-kB活化的抑制劑和TNFRSF1A接頭TRADD的低表達(圖4A和4B),以及mRNA水平的TNFRSF1B的高表達,表明Treg細胞在TNFR信號傳導中表現出適應性。脂質磷酸酶INPP5D(SHIP-1)的高表達抑制PI3K-AKT信號傳導,mTOR活化劑RPS6KA1和RPS6KA3的低表達,同樣也表明PI3K / AKT / mTOR途徑的適應性,Treg細胞中STAT4和NFATc2的低表達突出。為了確定上文鑒定的蛋白質表達模式的穩定性,作者在體外T細胞擴增后進行蛋白質組學分析。通過CD3、CD28和IL-2刺激將nTreg和nTconv細胞在體外擴增2周,孵育4天,并通過FOXP3、Helios、CTLA-4和CD25染色以及FOXP3基因TSDR甲基化驗證它們的身份。值得注意的是,即使在通過CD3和CD28途徑重新激活后,Treg細胞核心特征中大多數蛋白質特異性表達模式也基本上是保守的(圖4C,4D)。作者認為Treg細胞在某些代謝功能以及重要信號通路的組成中與Tconv細胞本質上不同。 圖4 常見的Treg細胞蛋白組學特征5. eTreg細胞特有的蛋白質組學特征
圖4 常見的Treg細胞蛋白組學特征5. eTreg細胞特有的蛋白質組學特征
除了常見的Treg細胞特征外,作者發現eTreg細胞與其他CD4+ T細胞亞群相比有獨特的蛋白質簇表達特征:具有相對高的(eTreghi)和低的(eTreglo)表達(圖5A)。eTreghi簇包括參與DNA復制(MCM2,-3,-4,-6,-7,FEN1)、有絲分裂(CORO1C,TUBA1B,TUBB,MYH9)(圖5A和5B)和細胞凋亡(FAS,CASP3)的蛋白質,這和已有研究一致,說明這些細胞正在發生分裂。在eTreghi簇中的41種蛋白質中,17種在體外Treg細胞擴增后仍顯示出上升趨勢,其中5種達到統計學顯著性。同樣,eTreglo簇中39種蛋白中的25種在體外擴增的Treg細胞中保持低表達。eTreglo簇包含多種調節細胞凋亡敏感性的GIMAP(圖5A和5B)。重要的是,eTreglo簇中幾乎40%的蛋白質在細胞信號傳導中具有功能,它們包括NF-kB途徑的多種成分(PRKCB、DPP4、NF-kB1、NF-kB2)、細胞因子受體途徑(IL-7R、INPP4B、STAM2、STAT3)和TCR途徑(TRAT1、VAV1、THEMIS)。即使在體外培養之后,Treg細胞中許多蛋白質的表達相對較低,表明這種對信號通路的明顯脫敏作用,并不僅僅是通過這些通路在體內激活信號的負面反饋調節。
FOXP3可以與關鍵轉錄因子(如IL17A和IL4)發生物理相互作用并且具有可以淬滅它們反式激活Tconv細胞效應基因的能力。反之亦然,如YY1的因子可以抑制FOXP3介導的Treg細胞程序的控制。作者為了確定Treg細胞中FOXP3的相對濃度是否足以“壓倒”這樣的相互作用因子,使用蛋白質組學標尺方法確定了每個細胞的蛋白質拷貝數,發現FOXP3在eTreg細胞中的拷貝數超過其許多伴侶蛋白,其中差別最大的是轉錄因子,如NFATc1和STAT4(圖5 C)。然而,對于一些轉錄因子(YY1、RUNX1和NFATc2),FOXP3過量僅為2至3倍,這可以解釋為什么FOXP3的適度減少允許Treg細胞產生效應細胞因子。最后,作者發現mTconv細胞中FOXP3的表達水平可能不足以有效中和其伴侶轉錄因子(圖5C)。 圖5 eTreg細胞蛋白特征6. eTreg細胞具有效應基因表達的遲鈍途徑
圖5 eTreg細胞蛋白特征6. eTreg細胞具有效應基因表達的遲鈍途徑
作者發現與Tconv細胞相比,eTreg細胞的IPA分析顯示TCR、模式識別受體(PRR)、細胞因子受體和TNFRSF家族誘導的NF-κB途徑信號傳導具有適應性。此外,通過NF-kB1和NF的流式細胞術證實,eTreg細胞中NF-kB1(p50)、NF-kB2(p52)和RELA(p65)的mRNA水平較低。因此,在eTreg細胞中,NF-kB1的反cd3和反cd28誘發的核核易位被削弱了,但在nTreg細胞中卻沒有。NFATc1和NFATc2蛋白水平在nTreg細胞和eTreg細胞中都較低。此外,NFATc2在普通Treg細胞的蛋白中含量較低,在Treg細胞在體外(圖4A和4D)后保持低表達。最后,在eTreg細胞轉錄組(圖6A)中,帶有NFATc2綁定元素的基因含量降低(圖6A),這表明該因素在體內的這些細胞中不那么活躍。炎性細胞因子控制Treg細胞行為可能會對其穩定性造成影響,在傳遞細胞因子受體信號的STAT轉錄因子中,STAT3、STAT6,尤其是STAT4在eTreg細胞中表現出低表達。STAT4表達在mRNA和蛋白質水平均較低,STAT4的低蛋白質表達是常見Treg細胞蛋白特征的一部分。在eTreg細胞轉錄組中,帶有STAT4結合元素的基因也沒有得到充分的表達(圖6A),這支持了體內eTreg細胞中STAT4活性降低的觀點。通過響應IL-12和I型IFN磷酸化Y693,在人CD4 + T細胞中激活STAT4,后者在人CD4 + Tconv細胞中誘導最強的早期STAT4磷酸化。這些細胞因子一致地誘導Treg細胞中STAT4的磷酸化比從血液中新鮮分離的Tconv細胞中更少(圖6C),并且這種差異在體外擴增的細胞中甚至更為顯著(圖6E)。STAT4是IFNg基因表達的主要調節因子,因此,作者推斷STAT4的低表達可能使Treg細胞與炎性細胞因子誘導IFN-g隔離。與此假設一致,IFN-α并且IL-12不能在Treg細胞中誘導IFN-g產生(圖6F)。然而,當故意過表達STAT4時,這些細胞因子確實激發了IFN-g的產生Treg細胞(圖6G)。值得注意的是,尤其是當用IFN-α或IL-12刺激Treg細胞時,STAT4的過表達也誘導了IL-2的產生(圖6G)和FOXP3表達的顯著喪失(圖6H和6I)。這些發現表明STAT4的低表達有助于在炎性環境中保護Treg細胞。
因為Treg細胞需要來自炎性細胞因子的輸入,所以雖然STAT4表達低,但是作者仍驗證了Treg細胞是否能夠對I型IFN起反應。結果發現,在Treg和Tconv細胞中均等地誘導STAT1磷酸化(圖6B和6D)。 此外,IFN-α容易在抗CD3和抗CD28活化的Treg細胞中誘導轉錄因子T-bet和趨化因子受體CXCR3的表達(圖6J,6K)。 總之,這些發現表明STAT4的選擇性低表達允許Treg細胞響應炎性細胞因子(例如歸巢至發炎組織),而不損害Treg細胞。 圖6 Treg細胞中選擇性缺乏STAT4激活7. 在Fr.III細胞中可以區分不同的細胞群
圖6 Treg細胞中選擇性缺乏STAT4激活7. 在Fr.III細胞中可以區分不同的細胞群
Fr.III細胞是CD4+CD25+并且表達FOXP3,可以??產生炎性細胞因子并且在體外缺乏強大抑制能力的細胞。該群體中的許多細胞表達CD127表明它是含有類似Treg或Tconv細胞的細胞的混合群體。為了更好地表征這一群體,作者根據CD127是否表達將其分成兩個部分,然后進行每個級分的蛋白質組學分析,并與nTconv、mTconv、nTreg和eTreg細胞的蛋白質組一起進行分析。層次聚類分析和PCA顯示CD127+ 亞群與mTconv細胞密切相關(圖7A和7B),說明CD127用于區分Tconv和Treg細胞有用性。值得注意的是,CD127- 亞群與eTreg細胞幾乎無法區分。CD127- Fr.III和eTreg細胞之間的緊密蛋白質組關系表明前者可能是真正的Treg細胞群,然而,FOXP3中的TSDR在CD127- Fr.III細胞中比在eTreg細胞中更加甲基化(圖7C)。此外,CD127-Fr.III細胞含有大部分產生一種或多種效應細胞因子的細胞(圖7F),這與eTreg細胞不同,因為eTreg細胞大多缺乏這種能力。并非CD127- Fr.III群體中的所有細胞都產生效應細胞因子,也并非eTreg細胞群中的所有細胞都缺乏產生此類細胞因子的能力,這表明即使這些明確定義的細胞群體仍可能是異質的。因此,作者在蛋白質組數據集中搜索了可能有助于區分具有不同功能特性的細胞的標記物。之前已有研究報道,在Treg細胞上發現的兩種標記物與陽性(CD49d)或陰性(CCR4)標志物與群體產生效應細胞因子的相對能力相關(圖7D)。流式細胞術分析顯示這些標志物表現出互斥的表達模式(圖7E),eTreg細胞群中產生IL-2或IL-17的少數細胞在表達CD49d的細胞中最為突出,這和已有的報道結果一致。然而,僅CD49d-CCR4 +表型完全排除了完全產生效應細胞因子的eTreg細胞(圖7G)。在CD127-Fr.III群體中觀察到類似的趨勢,其中CD49d-CCR4 +群體中不存在產生IL-17和IFN-g的細胞。然而,該群體在產生IL-2的能力方面仍然不同于相應的CD49d-CCR4 + eTreg細胞亞群(圖7G)。FOXP3的相對蛋白水平與每個群體中產生細胞因子的細胞的比例成反比。因此,FOXP3表達逐漸從eTreg細胞(最高)降低至Fr.III CD127-和Fr.III CD127 +(最低,但仍然高于mTconv細胞)。此外,在每個群體中,CCR4+CD49d-細胞總是表達比CCR4-CD49d +細胞更高的FOXP3。最后,TSDR僅在eTreg細胞中完全去甲基化,并且主要在CD127-CCR4+CD49d-Fr.III細胞中甲基化,其僅產生IL-2,但不產生其他炎性細胞因子。因此,作者的蛋白質組學數據使其發現CCR4和CD49d區分具有不同能力的細胞,以在eTreg細胞和Fr.III群體內產生效應細胞因子。這些標記物與常用的Treg細胞標記物組合的組合可用于細胞純化和診斷目的。例如,它們有助于闡明腫瘤中FOXP3+ CD4+T細胞的存在與患者愈后之間的關聯,特別是對于癌癥如結腸癌等。 文章小結
文章小結
通過對人類調節和常規CD4+T(Tconv)細胞的各種群體進行蛋白質組學和轉錄組學,獲得調節性T(Treg)細胞特性的分子特征,鑒定并定義了所有Treg細胞的蛋白質表達特征,以及定義效應Treg細胞的獨特特征。發現了Treg細胞中的代謝特征,以及細胞因子、TCR和共刺激受體信號傳導途徑的特異性適應性:適應——選擇性STAT4缺陷——保護Treg細胞穩態模型,該途徑通過炎性細胞因子起作用,而這些信號仍然可以通過其他途徑誘發關鍵的轉錄因子和引導受體。此外,作者的研究揭示了識別具有不同功能特性的FOXP3+ CD4+T細胞的表面標志物。研究結果表明,信號通路中的適應性保護Treg細胞,并為進一步研究Treg細胞生物學提供了資源。
參考文獻
1. Vogel, C., and Marcotte, E.M. (2012). Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 13, 227–232.
2. Arpaia, N., Green, J.A., et al . (2015). A distinct function of regulatory T cells in tissue protection. Cell 162, 1078–1089.
3. De Rosa, V., Galgani, M., et al. (2015). Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat. Immunol16, 1174–1184.
4. Oh, H., Grinberg-Bleyer, Y., et al. (2017). An NF-kB transcription-factor-dependent lineage-specific transcriptional program promotes regulatory T cell identity and function. Immunity 47, 450–465.e5.利用iTRAQ研究光控細菌代謝用于腫瘤治療
2018-06-25 16:56:45
題目:Optically-controlled bacterial metabolite for cancer therapy. Nature Communications IF 12.124 iTRAQ研究背景隨著對共生微生物認識的日益加深,人們逐漸意識到腫瘤內部是細菌和細胞共生的復雜微環境,并且相當一部分的細菌可以特異性地從循環系統中富集到腫瘤部位。在先前的研究中,科研人員也發現這些共生細菌在腫瘤的發展過程中起到關鍵性作用。在人為改造下,這一類共生細菌能夠富集到腫瘤部位,抑制腫瘤生長,從而為腫瘤治療提供全新方案。近來,許多納米級光催化材料如CdS和C3N4因其光電轉換能力引起了廣泛的關注,其可以將光能不斷轉換成電能。這些材料不僅能夠增加細菌的代謝活性,而且能夠避開負載極限,實現抗癌藥物的富產。受上述光誘導還原的啟發,本文作者將光催化系統與腫瘤靶向細菌融合,從而獲得用于光控制的一氧化氮(NO)生成的生物/非生物混合物。首先合成了抑制自由基生成能力的碳量子點摻雜的碳化氮(CCN),以實現原位光電轉換。然后,通過靜電相互作用組裝CCN和大腸桿菌(E. coli)以獲得CCN@E. coli。本文中,作者提出了一個利用修飾的CCN@E. coli的光控細菌代謝物療法(PMT)的概念。該技術利用修飾的CCN@E. coli將NO3-轉化為在光照下具有癌癥治療效果的抗腫瘤一氧化氮(NO)。作者還利用iTRAQ定量蛋白質組學技術來研究其詳細機制。PMT療法優化了生物/非生物混合系統在生物醫學中應用,轉變了細菌癌癥治療方式。研究內容及結果1. PMT系統的特性和機理CCN@E. coli的制備如圖1a所示。CCN通過靜電相互作用與E. coli組裝到一起。TEM圖像顯示修飾后,CCNs位于E. coli表面,在此過程中E. coli的形態沒有發生變化(圖1c)。如圖1d所示,CCN和E. coli之間的空間重疊系數(0.94)也表明CCN@E. coli組裝成功。作者推測,由于其合適的帶隙,從CCN激發的光電子能夠通過電子載體(例如NADH)轉移至大腸桿菌的NO合成酶中。隨后,NO3-可能以NADH依賴性方式酶促還原為NO(圖1g)。因此作者利用經典的Griess方法研究了CCN@E. coli光學控制的NO釋放行為(圖2a)。在正常生理條件下,L-精氨酸通過一氧化氮合成酶陽性細胞如巨噬細胞轉化為NO。然而,由于來源有限,并且NO可能最終被氧化成無毒的NO3-,所以生理條件下產生的NO的抗癌潛能力很微弱。在CCN@E.coli輔助下的PMT療法可將不可逆的腫瘤內NO代謝轉化為循環反應,最大限度地提高NO的生物利用度。PMT系統的機制是一個兩步過程:首先,E.coli利用發光的CCN光電產生的電子進行內源性NO3 -還原和NO生成(圖2g),然后,由產生的NO引發細胞凋亡。 2. PMT的體外抗癌研究為了使癌細胞和細菌在不直接接觸的情況下實現代謝物的交換,作者使用3D打印技術制作了一種共培養裝置。在這種培養體系中,NO可以通過CCN@E. coli(裝置外培養)的多孔膜擴散到癌細胞(裝置內培養),而CCN@E. coli或癌細胞都不能通過這個膜遷移。然后利用二氨基熒光素-FM二乙酸酯(DAF-FM DA)測定細胞內NO的濃度。如圖2i所示,4T1細胞中綠色熒光的增加表明,PMT系統產生的NO能有效擴散到附近的腔室,上調癌細胞中的NO的水平。沒有光照射的野生型大腸桿菌和CCN@E. coli不能使細胞內NO濃度增加(圖2j)。如圖2k所示,使用CCN@E. coli光輻射的細胞存活率顯著降低,在CCN@E. coli光輻射量為108 CFU/mL時,高達70%的4T1細胞能在24小時內被殺死。此外,作者還發現血紅蛋白(一種NO清除劑)和Ac-DEVD-CHO(一種凋亡抑制劑)能有效地阻礙4T1細胞的凋亡(圖2I)。基于上述觀察,作者推斷NO誘導的細胞凋亡是4T1細胞死亡的主要原因。
2. PMT的體外抗癌研究為了使癌細胞和細菌在不直接接觸的情況下實現代謝物的交換,作者使用3D打印技術制作了一種共培養裝置。在這種培養體系中,NO可以通過CCN@E. coli(裝置外培養)的多孔膜擴散到癌細胞(裝置內培養),而CCN@E. coli或癌細胞都不能通過這個膜遷移。然后利用二氨基熒光素-FM二乙酸酯(DAF-FM DA)測定細胞內NO的濃度。如圖2i所示,4T1細胞中綠色熒光的增加表明,PMT系統產生的NO能有效擴散到附近的腔室,上調癌細胞中的NO的水平。沒有光照射的野生型大腸桿菌和CCN@E. coli不能使細胞內NO濃度增加(圖2j)。如圖2k所示,使用CCN@E. coli光輻射的細胞存活率顯著降低,在CCN@E. coli光輻射量為108 CFU/mL時,高達70%的4T1細胞能在24小時內被殺死。此外,作者還發現血紅蛋白(一種NO清除劑)和Ac-DEVD-CHO(一種凋亡抑制劑)能有效地阻礙4T1細胞的凋亡(圖2I)。基于上述觀察,作者推斷NO誘導的細胞凋亡是4T1細胞死亡的主要原因。 3. PMT系統的體內生物分布研究為評估PMT系統的腫瘤靶向能力,將DIR標記的CCN@E.coli靜脈注射到4T1荷瘤小鼠。如圖3a所示,隨著時間的延長,在腫瘤位置內檢測到熒光的也逐漸增強。如圖3b所示,離體熒光成像顯示大量的細菌積聚在腫瘤位置,而肝臟或腎臟滯留的細菌幾乎可忽略不計。值得注意的是,CCN的修飾并不影響E.coli的腫瘤靶向能力(圖3c)。通常,大多數合成藥物載體由于其擴散限制,幾乎不能穿透腫瘤組織。而作者推測PMT系統可能能夠到達移植瘤的較深區域。結果表明,不同深度的腫瘤組織切片顯示PMT系統分布于腫瘤組織中(圖3d),并且發現移植瘤的中心聚積的細菌更多。為了全面揭示CCN@E.coli的腫瘤穿透性特征。作者使用表面活性劑輔助組織清除技術,即CLARITY,將腫瘤組織轉化為透明的形式(圖3e)。然后使用與腫瘤缺氧高度相關的生物標志物碳酸酐酶-Ⅸ(CA-9)對腫瘤缺氧區進行染色。從3D腫瘤熒光圖像可觀察到來自CCN@E.coli的紅色熒光和來自CA-9的綠色熒光的共定位,而CCN@E.coli熒光可以在含氧量正常的區域發現(圖3f)。進而證明了PMT系統可以充分利用大腸桿菌缺氧介導的趨化性,并且達到其他常規載體無法高效利用的缺氧區域。隨后,作者研究了CCN@E.coli在主要代謝器官如肝、脾和腎中的清除動力學。通過PCR和定性免疫熒光染色證明大部分細菌可以隨著的時間延長而被清除。3周后,在免疫活性小鼠中可以檢測到極少量的細菌(圖3h),血液生化和血液學分析未發現PMT的長期副作用(圖3i)。
3. PMT系統的體內生物分布研究為評估PMT系統的腫瘤靶向能力,將DIR標記的CCN@E.coli靜脈注射到4T1荷瘤小鼠。如圖3a所示,隨著時間的延長,在腫瘤位置內檢測到熒光的也逐漸增強。如圖3b所示,離體熒光成像顯示大量的細菌積聚在腫瘤位置,而肝臟或腎臟滯留的細菌幾乎可忽略不計。值得注意的是,CCN的修飾并不影響E.coli的腫瘤靶向能力(圖3c)。通常,大多數合成藥物載體由于其擴散限制,幾乎不能穿透腫瘤組織。而作者推測PMT系統可能能夠到達移植瘤的較深區域。結果表明,不同深度的腫瘤組織切片顯示PMT系統分布于腫瘤組織中(圖3d),并且發現移植瘤的中心聚積的細菌更多。為了全面揭示CCN@E.coli的腫瘤穿透性特征。作者使用表面活性劑輔助組織清除技術,即CLARITY,將腫瘤組織轉化為透明的形式(圖3e)。然后使用與腫瘤缺氧高度相關的生物標志物碳酸酐酶-Ⅸ(CA-9)對腫瘤缺氧區進行染色。從3D腫瘤熒光圖像可觀察到來自CCN@E.coli的紅色熒光和來自CA-9的綠色熒光的共定位,而CCN@E.coli熒光可以在含氧量正常的區域發現(圖3f)。進而證明了PMT系統可以充分利用大腸桿菌缺氧介導的趨化性,并且達到其他常規載體無法高效利用的缺氧區域。隨后,作者研究了CCN@E.coli在主要代謝器官如肝、脾和腎中的清除動力學。通過PCR和定性免疫熒光染色證明大部分細菌可以隨著的時間延長而被清除。3周后,在免疫活性小鼠中可以檢測到極少量的細菌(圖3h),血液生化和血液學分析未發現PMT的長期副作用(圖3i)。 4. PMT系統中體內NO的生成為了進一步闡明細胞光合行為,作者設計并構建了Nrf2受控螢光素酶表達質粒用以探測體內NO的產生,之后轉染4T1細胞以構建4T1Nrf2細胞用于原位NO的檢測。如圖4a所示,4T1Nrf2-luc細胞暴露在CCN@E.coli環境下會導致其生物發光快速增加,作者還研究了4T1Nrf2-luc腫瘤小鼠中PMT系統的體內效率。如圖4b所示,單獨的CCN和E.coli都不能誘導產生可檢測的NO。而CCN@E.coli處理組的生物發光強度顯著增加。接下來,作者還合成了用于NO檢測的磁共振成像(MRI)探針Fe-MGD。如圖4c所示,作者發現在注射了CCN@E.coli和光照后,小鼠腫瘤內的T1信號增強,進一步證明了體內光控制的NO生成。
4. PMT系統中體內NO的生成為了進一步闡明細胞光合行為,作者設計并構建了Nrf2受控螢光素酶表達質粒用以探測體內NO的產生,之后轉染4T1細胞以構建4T1Nrf2細胞用于原位NO的檢測。如圖4a所示,4T1Nrf2-luc細胞暴露在CCN@E.coli環境下會導致其生物發光快速增加,作者還研究了4T1Nrf2-luc腫瘤小鼠中PMT系統的體內效率。如圖4b所示,單獨的CCN和E.coli都不能誘導產生可檢測的NO。而CCN@E.coli處理組的生物發光強度顯著增加。接下來,作者還合成了用于NO檢測的磁共振成像(MRI)探針Fe-MGD。如圖4c所示,作者發現在注射了CCN@E.coli和光照后,小鼠腫瘤內的T1信號增強,進一步證明了體內光控制的NO生成。 5. PMT的體內抗癌機制作者利用綴合有Cy5.5的膜聯蛋白V29來研究PMT的體內治療反應。如圖4d所示,隨著CCN@E.coli注射劑量的增加,CCN@E.coli在腫瘤部位的累積增多。圖4e的半定量分析結果也證明了腫瘤內細菌數目、腫瘤細胞毒性和NO濃度呈正相關。此外,作者在4T1荷瘤小鼠和CT26荷瘤小鼠中檢查了PMT的抗癌效率,治療時間表如圖4f所示。在4T1荷瘤小鼠中,PMT治療抑制了79.3%的腫瘤生長(圖4g)。而在CT26荷瘤小鼠中,PMT治療抑制了70.2%的腫瘤生長(圖4h)。這些結果表明PMT應該能夠適用于治療不同類型的癌癥。隨后,作者利用定量蛋白質組學技術探索了PMT的詳細抗癌機制。利用iTRAQ技術從4T1荷瘤小鼠癌組織(PMT處理組和PBS對照組)中共鑒定到4735個蛋白。如圖5a所示,在PMT治療后,腫瘤組織內鑒定到222個上調的差異蛋白和17個下調的差異蛋白(倍數變化≥1.5和P <0.05,超幾何檢驗)。聚類分析和主成分分析結果顯示PBS處理組和PMT處理組之間存在顯著差異。對差異蛋白進行GO分析發現,刺激應答、信號、細胞死亡、免疫系統和細胞殺傷相關的蛋白質在PMT組中顯著上調(圖5b)。相反,與細胞增殖和生長相關的蛋白更多地發生下調。這些數據表明PMT在腫瘤中引起高應激反應,這主要是由于NO誘導的氧化損傷,并且與抗氧化活性相關的蛋白質也顯著降低。通常,NO對癌細胞的細胞毒性主要歸因于其誘導氧化應激和引發DNA損傷的能力。圖5c所示的維恩圖表明,大腸桿菌感染/侵入相關蛋白與DNA損傷/氧化應激相關蛋白之間沒有重疊。因此,蛋白質組學研究表明CCN@E.coli對癌細胞的直接細胞毒性主要歸因于NO本身。有趣的是,在GO分析中發現了參與免疫應答的蛋白質水平顯著升高。這一現象表明免疫系統可能有助于體內PMT的抗癌作用。PMT治療后蛋白相互作用網絡分析也可得出同樣的結論。如圖5e所示,KEGG分析發現抗原遞呈途徑被全面激活,并且證實了HMGB蛋白(一種參與觸發抗原呈遞的主導蛋白導致免疫原性細胞死亡)的上調。這些發現揭示了PMT可能通過HMGB引發MHC I類介導的途徑誘導免疫原性細胞死亡。在體外模擬環境中,發現PMT治療能夠誘導DC突變(圖5f)。這些結果表明,除了直接產生細胞毒性NO外,免疫應答也可能在在PMT的抗癌機制中起關鍵作用。
5. PMT的體內抗癌機制作者利用綴合有Cy5.5的膜聯蛋白V29來研究PMT的體內治療反應。如圖4d所示,隨著CCN@E.coli注射劑量的增加,CCN@E.coli在腫瘤部位的累積增多。圖4e的半定量分析結果也證明了腫瘤內細菌數目、腫瘤細胞毒性和NO濃度呈正相關。此外,作者在4T1荷瘤小鼠和CT26荷瘤小鼠中檢查了PMT的抗癌效率,治療時間表如圖4f所示。在4T1荷瘤小鼠中,PMT治療抑制了79.3%的腫瘤生長(圖4g)。而在CT26荷瘤小鼠中,PMT治療抑制了70.2%的腫瘤生長(圖4h)。這些結果表明PMT應該能夠適用于治療不同類型的癌癥。隨后,作者利用定量蛋白質組學技術探索了PMT的詳細抗癌機制。利用iTRAQ技術從4T1荷瘤小鼠癌組織(PMT處理組和PBS對照組)中共鑒定到4735個蛋白。如圖5a所示,在PMT治療后,腫瘤組織內鑒定到222個上調的差異蛋白和17個下調的差異蛋白(倍數變化≥1.5和P <0.05,超幾何檢驗)。聚類分析和主成分分析結果顯示PBS處理組和PMT處理組之間存在顯著差異。對差異蛋白進行GO分析發現,刺激應答、信號、細胞死亡、免疫系統和細胞殺傷相關的蛋白質在PMT組中顯著上調(圖5b)。相反,與細胞增殖和生長相關的蛋白更多地發生下調。這些數據表明PMT在腫瘤中引起高應激反應,這主要是由于NO誘導的氧化損傷,并且與抗氧化活性相關的蛋白質也顯著降低。通常,NO對癌細胞的細胞毒性主要歸因于其誘導氧化應激和引發DNA損傷的能力。圖5c所示的維恩圖表明,大腸桿菌感染/侵入相關蛋白與DNA損傷/氧化應激相關蛋白之間沒有重疊。因此,蛋白質組學研究表明CCN@E.coli對癌細胞的直接細胞毒性主要歸因于NO本身。有趣的是,在GO分析中發現了參與免疫應答的蛋白質水平顯著升高。這一現象表明免疫系統可能有助于體內PMT的抗癌作用。PMT治療后蛋白相互作用網絡分析也可得出同樣的結論。如圖5e所示,KEGG分析發現抗原遞呈途徑被全面激活,并且證實了HMGB蛋白(一種參與觸發抗原呈遞的主導蛋白導致免疫原性細胞死亡)的上調。這些發現揭示了PMT可能通過HMGB引發MHC I類介導的途徑誘導免疫原性細胞死亡。在體外模擬環境中,發現PMT治療能夠誘導DC突變(圖5f)。這些結果表明,除了直接產生細胞毒性NO外,免疫應答也可能在在PMT的抗癌機制中起關鍵作用。 文章小結一氧化氮(NO)在濃度較高的環境可以引發腫瘤細胞凋亡。為了實現對細菌合成NO能力的提升及控制,作者將碳量子點摻雜的碳化氮(CCN)負載到大腸桿菌(E.coli)MG1655上,利用合成材料良好的光催化性能,從而提高NO的產生,這一方法可以很好地實現細菌在腫瘤富集。本文作者全面研究了光控細菌代謝療法(PMT)中NO的生成,細胞毒性細胞殺傷作用及其相關機制。動物實驗證明,該療法在小鼠腫瘤模型上表現出了約80%的抑瘤率。這一發現對哺乳動物-微生物的共生關系有了更深認識,并且極大豐富現有腫瘤療法的內涵。定量蛋白質組學結果也表明免疫反應可能與該療法相關, PMT療法可能對癌癥免疫療法具有促進作用。該研究策略將為活體生物材料的設計和制備提供一種新的方法和視角。參考文獻1. Treweek, J. B. et al. Whole-body tissue stabilization and selective extractions via tissue-hydrogel hybrids for high-resolution intact circuit mapping and phenotyping. Nat. Protoc. 10, 1860–1896 (2015).2. Luo, C. H., Huang, C. T., Su, C. H. & Yeh, C. S. Bacteria-mediated hypoxiaspecific delivery of nanoparticles for tumors imaging and therapy. Nano Lett.16, 3493–3499 (2016).3. Zheng, J. H. et al. Two-step enhanced cancer immunotherapy with engineered Salmonella typhimurium secreting heterologous flagellin. Sci. Transl. Med. 9, eaak9537 (2016).4. Zitvogel, L., Daillere, R., Roberti, M. P., Routy, B. & Kroemer, G. Anticancer effects of the microbiome and its products. Nat. Rev. Microbiol. 15, 465–478 (2017).5. Wang, B. et al. Enhanced biological hydrogen production from Escherichia coli with surface precipitated cadmium sulfide nanoparticles. Adv. Energy Mater. 7, 1700611 (2017).
文章小結一氧化氮(NO)在濃度較高的環境可以引發腫瘤細胞凋亡。為了實現對細菌合成NO能力的提升及控制,作者將碳量子點摻雜的碳化氮(CCN)負載到大腸桿菌(E.coli)MG1655上,利用合成材料良好的光催化性能,從而提高NO的產生,這一方法可以很好地實現細菌在腫瘤富集。本文作者全面研究了光控細菌代謝療法(PMT)中NO的生成,細胞毒性細胞殺傷作用及其相關機制。動物實驗證明,該療法在小鼠腫瘤模型上表現出了約80%的抑瘤率。這一發現對哺乳動物-微生物的共生關系有了更深認識,并且極大豐富現有腫瘤療法的內涵。定量蛋白質組學結果也表明免疫反應可能與該療法相關, PMT療法可能對癌癥免疫療法具有促進作用。該研究策略將為活體生物材料的設計和制備提供一種新的方法和視角。參考文獻1. Treweek, J. B. et al. Whole-body tissue stabilization and selective extractions via tissue-hydrogel hybrids for high-resolution intact circuit mapping and phenotyping. Nat. Protoc. 10, 1860–1896 (2015).2. Luo, C. H., Huang, C. T., Su, C. H. & Yeh, C. S. Bacteria-mediated hypoxiaspecific delivery of nanoparticles for tumors imaging and therapy. Nano Lett.16, 3493–3499 (2016).3. Zheng, J. H. et al. Two-step enhanced cancer immunotherapy with engineered Salmonella typhimurium secreting heterologous flagellin. Sci. Transl. Med. 9, eaak9537 (2016).4. Zitvogel, L., Daillere, R., Roberti, M. P., Routy, B. & Kroemer, G. Anticancer effects of the microbiome and its products. Nat. Rev. Microbiol. 15, 465–478 (2017).5. Wang, B. et al. Enhanced biological hydrogen production from Escherichia coli with surface precipitated cadmium sulfide nanoparticles. Adv. Energy Mater. 7, 1700611 (2017).iTRAQ蛋白質組揭示油菜卷葉機理
2018-06-25 16:18:55
文獻:Histological, Physiological, and Comparative Proteomic Analyses Provide Insights into Leaf Rolling in Brassica napus.Journal of Proteome Research IF4.268 iTRAQ研究背景葉片是大多數植物光合作用的主要器官,葉片發育影響作物產量和植物結構。適度的卷葉被認為是作物理想型育種的一個重要組成部分,它能夠改變植物的結構,提高光合效率,延緩葉片衰老,減輕干旱、高溫和高光等脅迫帶來的損害。近年來,在擬南芥和水稻等物種中,已經分離出了卷曲葉片突變體,并對它們的分子機制進行了深入的研究。油菜作為世界上最重要的油料作物之一,雖然也發現了一些卷曲葉片的突變體,但是對于其作用機制仍有待進一步的研究。本文作者主要利用iTRAQ定量蛋白質組學技術分析油菜卷葉突變株Bndcl1與野生株(WT)的差異蛋白,闡明了卷葉機理及其對植物的生理影響,有望促進油菜的理想株型育種。研究內容及結果1. 本研究中,作者選取了油菜卷葉突變株Bndcl1,首先經組織學對比發現,相比于野生株(WT),突變株(Bndcl1)幼苗期的葉片表型上比較緊湊(圖 1A和B)。組織切片結果中發現Bndcl1葉綠體數目明顯增多,特別是在海綿狀的葉肉細胞中(圖1C和D)。此外,作者發現,在Bndcl1突變體中,在遠軸端韌皮部組織細胞較少,而在其葉脈以下的遠軸端表皮細胞明顯增大(圖1E和F)。 2. 為了評估卷曲葉片對Bndcl1突變株光合性能的影響,作者測定了Chl的含量、Chl熒光和氣體交換參數。與野生株相比,Bndcl1株中的葉綠體數量、PSII和凈光合速率的有效量子產量顯著增加。3. 活性氧(ROS)的生成在光合作用的光反應過程中是不可避免的,PSII極易受到光破壞的影響。光抑制的光合作用不可避免的損失會因ROS的產生而加劇,而ROS的產生會減慢PSII的修復。為了減少光氧化傷害,光合生物體已經進化出多種機制,而對抗氧化劑的上調是調節機制的一部分。因此,作者檢測了Bndcl1和WT葉子的酶活性、脂質過氧化和ROS水平,發現在Bndcl1葉片中O2? ? 和H2O2較低,由此說明其ROS水平也將明顯降低(圖2A和B)。此外,TBARS在突變株的低水平同樣說明其膜脂過氧化的水平降低(圖2C)。作者還檢測比較了兩者的SOD、POD和CAT酶活性,發現相比于WT株,在Bndcl1葉片中此酶活性顯著增加。由此說明Bndcl1的光系統能更好的避免受到光氧化傷害。
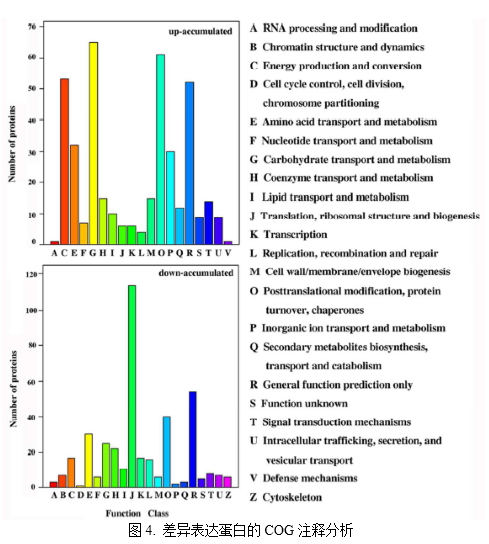
2. 為了評估卷曲葉片對Bndcl1突變株光合性能的影響,作者測定了Chl的含量、Chl熒光和氣體交換參數。與野生株相比,Bndcl1株中的葉綠體數量、PSII和凈光合速率的有效量子產量顯著增加。3. 活性氧(ROS)的生成在光合作用的光反應過程中是不可避免的,PSII極易受到光破壞的影響。光抑制的光合作用不可避免的損失會因ROS的產生而加劇,而ROS的產生會減慢PSII的修復。為了減少光氧化傷害,光合生物體已經進化出多種機制,而對抗氧化劑的上調是調節機制的一部分。因此,作者檢測了Bndcl1和WT葉子的酶活性、脂質過氧化和ROS水平,發現在Bndcl1葉片中O2? ? 和H2O2較低,由此說明其ROS水平也將明顯降低(圖2A和B)。此外,TBARS在突變株的低水平同樣說明其膜脂過氧化的水平降低(圖2C)。作者還檢測比較了兩者的SOD、POD和CAT酶活性,發現相比于WT株,在Bndcl1葉片中此酶活性顯著增加。由此說明Bndcl1的光系統能更好的避免受到光氧化傷害。 4. 作者應用iTRAQ定量蛋白質組學技術研究Bndcl1與WT葉片蛋白質表達水平的差異,總共鑒定到5019個蛋白,差異蛋白有943個,其中發生上調的蛋白有451個,發生下調的蛋白有492個。作者對這些蛋白進行了GO、COG、KEGG等注釋分析。通過對差異蛋白中發生上調和下調的蛋白分別進行GO分析并對比發現,在“細胞增殖”、“生物過程的調節”、“色素沉著”等功能方面,下調的蛋白數目要比上調的蛋白數多,具有抗氧化活性的功能的上調蛋白數目是下調蛋白數目的3倍多(圖3)。COG分析表明,大多數下調蛋白參與翻譯、核糖體結構和生物發生,而上調蛋白主要參與碳水化合物的運輸和代謝、轉錄翻譯后修飾、蛋白質的周轉、分子伴侶和能量的產生和轉化(圖4)。KEGG 注釋結果顯示,盡管在“代謝途徑”、“次生代謝產物的生物合成”和“淀粉和蔗糖代謝”中均有上調和下調的蛋白質參與,但在不同的途徑中也涉及到不同差異蛋白質(圖5)。
4. 作者應用iTRAQ定量蛋白質組學技術研究Bndcl1與WT葉片蛋白質表達水平的差異,總共鑒定到5019個蛋白,差異蛋白有943個,其中發生上調的蛋白有451個,發生下調的蛋白有492個。作者對這些蛋白進行了GO、COG、KEGG等注釋分析。通過對差異蛋白中發生上調和下調的蛋白分別進行GO分析并對比發現,在“細胞增殖”、“生物過程的調節”、“色素沉著”等功能方面,下調的蛋白數目要比上調的蛋白數多,具有抗氧化活性的功能的上調蛋白數目是下調蛋白數目的3倍多(圖3)。COG分析表明,大多數下調蛋白參與翻譯、核糖體結構和生物發生,而上調蛋白主要參與碳水化合物的運輸和代謝、轉錄翻譯后修飾、蛋白質的周轉、分子伴侶和能量的產生和轉化(圖4)。KEGG 注釋結果顯示,盡管在“代謝途徑”、“次生代謝產物的生物合成”和“淀粉和蔗糖代謝”中均有上調和下調的蛋白質參與,但在不同的途徑中也涉及到不同差異蛋白質(圖5)。
 5. 為了進一步對這些差異蛋白的功能進行分類分析,作者進行了功能富集分析。通過對差異蛋白的富集分析結果發現,這些發生上調的蛋白能夠改善Bndcl1株的光合性能和減輕氧化傷害,并且發現下調的蛋白參與葉片近-遠軸極性建成。6. 作者最后從差異表達蛋白中挑選了多個蛋白進行了qRT-PCR表達驗證。例如:與卷葉表型相關的基因PGY1, PGY2, PGY3, AE5, STV1 and SGS3,這幾個基因的蛋白表達水平在蛋白質組學結果是顯著下調。qRT-PCR驗證結果顯示,在Bndcl1株,這些基因mRNA表達水平顯著降低,表明這些基因在蛋白質和mRNA水平上發生了一致的變化(圖6A)。另外作者從轉錄水平上檢測了葉極性的關鍵調節因子:AS1, AS2, KAN1, KAN2, REV, RDR6, AGO7, AGO1(圖6B)。結果發現在Bndcl1株,AS1, KAN2發生了顯著降低,AS2, KAN1沒有顯著變化,REV發生了顯著下調。此外,RDR6, AGO7, AGO1也同樣發生了顯著下調。最后作者還對油菜甾醇相關的6個差異蛋白進行了轉錄水平的驗證,結果與iTRAQ數據(圖6C)一致,說明了定量蛋白質組學分析所獲得的結果的可靠性。
5. 為了進一步對這些差異蛋白的功能進行分類分析,作者進行了功能富集分析。通過對差異蛋白的富集分析結果發現,這些發生上調的蛋白能夠改善Bndcl1株的光合性能和減輕氧化傷害,并且發現下調的蛋白參與葉片近-遠軸極性建成。6. 作者最后從差異表達蛋白中挑選了多個蛋白進行了qRT-PCR表達驗證。例如:與卷葉表型相關的基因PGY1, PGY2, PGY3, AE5, STV1 and SGS3,這幾個基因的蛋白表達水平在蛋白質組學結果是顯著下調。qRT-PCR驗證結果顯示,在Bndcl1株,這些基因mRNA表達水平顯著降低,表明這些基因在蛋白質和mRNA水平上發生了一致的變化(圖6A)。另外作者從轉錄水平上檢測了葉極性的關鍵調節因子:AS1, AS2, KAN1, KAN2, REV, RDR6, AGO7, AGO1(圖6B)。結果發現在Bndcl1株,AS1, KAN2發生了顯著降低,AS2, KAN1沒有顯著變化,REV發生了顯著下調。此外,RDR6, AGO7, AGO1也同樣發生了顯著下調。最后作者還對油菜甾醇相關的6個差異蛋白進行了轉錄水平的驗證,結果與iTRAQ數據(圖6C)一致,說明了定量蛋白質組學分析所獲得的結果的可靠性。 文章小結這項結果研究表明,葉片近-遠軸極性和油菜甾醇代謝及信號通路的缺陷是導致Bndcl1突變株葉子向下卷曲表型的原因,葉片的適度卷曲能夠提高光吸收、能量轉移和CO2固定效率,進而提高了突變株的光合性能。此外,在突變株中,參與PSII修復周期的抗氧化劑和蛋白質水平的升高可能會減少非生物脅迫的光合損失。這項研究為油菜卷葉機制及卷葉對植物生理的影響提供了新的見解。因此,卷葉突變株因具有緊湊的結構,能提高光合效率和增強抗逆性,這也是作物育種理想株型。參考文獻1. Yang, C.; Xu, L.; et al. iTRAQ-based proteomics of sunflower cultivars differing in resistance to parasitic weed Orobanche cumana. Proteomics 2017, 17, 1700009.2. Liang, J.; Liu, B.; et al. Genetic variation and divergence of genes involved in leaf adaxial-abaxial polarity establishment in Brassica rapa. Front. Plant Sci. 2016, 7, 94.3. Husbands, A. Y.; Benkovics, A. H.; et al. The ASYMMETRIC LEAVES complex employs multiple modes of regulation to affect adaxial-abaxial patterning and leaf complexity. Plant Cell 2015, 27, 3321-3335.4. Jia, H.; Shao, M.; et al. Proteome dynamics and physiological responses to short-term salt stress in brassica napus leaves. Plos One 2015, 10, e0144808.5. Yamaguchi, T.; Nukazuka, A.; Tsukaya, H. Leaf adaxial-abaxial polarity specification and laminaoutgrowth: Evolution and development. Plant Cell Physiol. 2012, 53, 1180-1194.
文章小結這項結果研究表明,葉片近-遠軸極性和油菜甾醇代謝及信號通路的缺陷是導致Bndcl1突變株葉子向下卷曲表型的原因,葉片的適度卷曲能夠提高光吸收、能量轉移和CO2固定效率,進而提高了突變株的光合性能。此外,在突變株中,參與PSII修復周期的抗氧化劑和蛋白質水平的升高可能會減少非生物脅迫的光合損失。這項研究為油菜卷葉機制及卷葉對植物生理的影響提供了新的見解。因此,卷葉突變株因具有緊湊的結構,能提高光合效率和增強抗逆性,這也是作物育種理想株型。參考文獻1. Yang, C.; Xu, L.; et al. iTRAQ-based proteomics of sunflower cultivars differing in resistance to parasitic weed Orobanche cumana. Proteomics 2017, 17, 1700009.2. Liang, J.; Liu, B.; et al. Genetic variation and divergence of genes involved in leaf adaxial-abaxial polarity establishment in Brassica rapa. Front. Plant Sci. 2016, 7, 94.3. Husbands, A. Y.; Benkovics, A. H.; et al. The ASYMMETRIC LEAVES complex employs multiple modes of regulation to affect adaxial-abaxial patterning and leaf complexity. Plant Cell 2015, 27, 3321-3335.4. Jia, H.; Shao, M.; et al. Proteome dynamics and physiological responses to short-term salt stress in brassica napus leaves. Plos One 2015, 10, e0144808.5. Yamaguchi, T.; Nukazuka, A.; Tsukaya, H. Leaf adaxial-abaxial polarity specification and laminaoutgrowth: Evolution and development. Plant Cell Physiol. 2012, 53, 1180-1194.Nature Genetics丨三維基因組學研究進展
2018-05-04 14:13:14
4月26日,武漢金開瑞生物工程有限公司技術顧問、華中農業大學教授, 曹罡課題組與華中農業大學李國亮教授課題組在國際著名期刊Nature Genetics上聯合發表題為“Digestion-ligation-only Hi-C is an efficient andcost-effective method for chromosome conformation capture”的研究論文。該論文主要介紹了一種新的染色體構象捕獲技術Hi-C,為解析基因組三維結構提供了一種新型、高效、經濟的研究方法。 基因組三維空間結構與功能的研究簡稱三維基因組學(Three-Dimensional Genomics,3D Genomics),是指在考慮基因組序列、基因結構及其調控元件的同時,對基因組序列在細胞核內的三維空間結構,及其在基因轉錄、調控、復制和修復等生物過程中功能的研究。染色體是由DNA與組蛋白共同組成,從染色體的一級結構(繩珠模型)到四級超螺旋折疊結構,DNA分子一共被壓縮了8400倍左右,正是這些折疊和壓縮,導致基因在細胞中的分布復雜而又有序,只有了解清楚染色體區域(A/B compartments、TADs、Loops),才可以將基因組上原本分散的遠距離調控元件與其具體調控區域更好的關聯起來,對理解基因的轉錄調控、增強子與啟動子的相互作用、疾病易感位點、DNA損傷修復、基因組結構變異和表觀遺傳有著重要的意義。科學家們對染色體三維結構的研究很早就開始了,但是相關的研究一直不夠深入,同時缺乏微觀的證據。直到2003年Job Dekker及其合作者提出了染色質構象捕獲技術(Chromatin Conformation Capture,3C),用于測定特定的點到點之間的染色質交互作用。隨后,科學家們擴展了3C技術,開發了4C技術(Circularized Chromatin Conformation Capture),用于測定一點到多點之間的染色質交互作用。Dostie等人接著開發了5C技術(Carbon-Copy Chromatin Conformation Capture),用于測定多點到多點之間的染色質交互作用。為了能捕獲全基因組范圍的染色質相互作用,Job Dekker研究組又開發出了現在大家所熟知的Hi-C技術。Hi-C技術是三維基因組學的主要研究方法,但存在實驗成本高、數據噪音大、實驗過程繁瑣等缺點。為解決這些問題,曹罡教授及其合作者提供了一種新的DLO Hi-C染色體構象捕獲技術(digestion-ligation-only Hi-C , DLO Hi-C),設計了巧妙的酶切位點,采用同時酶切酶連的方式,將DNA接頭連接在染色體內切酶切口末端上,然后進行鄰近酶連,最后再用MmeI內切酶酶切消化,回收固定大小互作DNA片段,從而大大地降低了測序背景數據噪音,獲取的測序數據質量高于傳統Hi-C。DLO Hi-C技術還可以用于染色體易位位點篩選。
基因組三維空間結構與功能的研究簡稱三維基因組學(Three-Dimensional Genomics,3D Genomics),是指在考慮基因組序列、基因結構及其調控元件的同時,對基因組序列在細胞核內的三維空間結構,及其在基因轉錄、調控、復制和修復等生物過程中功能的研究。染色體是由DNA與組蛋白共同組成,從染色體的一級結構(繩珠模型)到四級超螺旋折疊結構,DNA分子一共被壓縮了8400倍左右,正是這些折疊和壓縮,導致基因在細胞中的分布復雜而又有序,只有了解清楚染色體區域(A/B compartments、TADs、Loops),才可以將基因組上原本分散的遠距離調控元件與其具體調控區域更好的關聯起來,對理解基因的轉錄調控、增強子與啟動子的相互作用、疾病易感位點、DNA損傷修復、基因組結構變異和表觀遺傳有著重要的意義。科學家們對染色體三維結構的研究很早就開始了,但是相關的研究一直不夠深入,同時缺乏微觀的證據。直到2003年Job Dekker及其合作者提出了染色質構象捕獲技術(Chromatin Conformation Capture,3C),用于測定特定的點到點之間的染色質交互作用。隨后,科學家們擴展了3C技術,開發了4C技術(Circularized Chromatin Conformation Capture),用于測定一點到多點之間的染色質交互作用。Dostie等人接著開發了5C技術(Carbon-Copy Chromatin Conformation Capture),用于測定多點到多點之間的染色質交互作用。為了能捕獲全基因組范圍的染色質相互作用,Job Dekker研究組又開發出了現在大家所熟知的Hi-C技術。Hi-C技術是三維基因組學的主要研究方法,但存在實驗成本高、數據噪音大、實驗過程繁瑣等缺點。為解決這些問題,曹罡教授及其合作者提供了一種新的DLO Hi-C染色體構象捕獲技術(digestion-ligation-only Hi-C , DLO Hi-C),設計了巧妙的酶切位點,采用同時酶切酶連的方式,將DNA接頭連接在染色體內切酶切口末端上,然后進行鄰近酶連,最后再用MmeI內切酶酶切消化,回收固定大小互作DNA片段,從而大大地降低了測序背景數據噪音,獲取的測序數據質量高于傳統Hi-C。DLO Hi-C技術還可以用于染色體易位位點篩選。 DLO Hi-C技術使全基因組染色體構象捕獲實驗的成本大大的降低,同時簡化了實驗步驟,使得實驗成功率顯著提高,對輔助基因組組裝、解析基因組遠程調控元件的功能、理解疾病易感位點以及檢測染色體結構變異有著重要的意義。Nature Genetics同期發表專門評論性文章,對曹罡教授及其合作者的研究工作給予了較高評價“Lin et al. introduce an elegant dual-linkerstrategy that allows for noise filtering and early quality control.”
DLO Hi-C技術使全基因組染色體構象捕獲實驗的成本大大的降低,同時簡化了實驗步驟,使得實驗成功率顯著提高,對輔助基因組組裝、解析基因組遠程調控元件的功能、理解疾病易感位點以及檢測染色體結構變異有著重要的意義。Nature Genetics同期發表專門評論性文章,對曹罡教授及其合作者的研究工作給予了較高評價“Lin et al. introduce an elegant dual-linkerstrategy that allows for noise filtering and early quality control.” 隨后還將詳細解讀該篇論文,從實驗設計、流程、生信分析及可視化方面進行詳細說明,干貨滿滿,敬情期待哦!文章鏈接:https://www.nature.com/articles/s41588-018-0111-2
隨后還將詳細解讀該篇論文,從實驗設計、流程、生信分析及可視化方面進行詳細說明,干貨滿滿,敬情期待哦!文章鏈接:https://www.nature.com/articles/s41588-018-0111-2番茄轉錄因子SlHZ24通過對D-甘露糖/L半乳糖通路的正調節 促進抗壞血酸積累
2018-04-28 09:35:52
文獻來源:The tomato HD-Zip I transcription factor SlHZ24 modulates ascorbate accumulation through positive regulation of the D-mannose/L-galactose pathway.The Plant Journal(IF5.901)關鍵技術:酵母單/雙雜交、雙螢光素酶報告基因檢測、EMSA番茄轉錄因子SlHZ24通過對D-甘露糖/L半乳糖通路的正調節促進抗壞血酸積累
研究背景抗壞血酸(AsA)一種抗氧化劑,可以清除植物在脅迫條件下產生的活性氧。本文通過酵母單雜交技術、瞬時表達和EMSA實驗驗證了SlHZ24和SIGMP3啟動子結合,且SlHZ24表達水平與AsA積累呈正相關特性。相反,通過RNAi技術導致AsA表達量降低。SlHZ24同時也影響D-甘露糖/L半乳糖通路其他基因的表達,如SIGME2、SIGGP和SIGMP4,說明抗壞血酸合成為多靶點調控。SlHZ24過表達植株對氧化應激的敏感性降低,作者推斷SlHZ24促進抗壞血酸的合成,提高了植株對氧化應激的耐受性。研究內容1.SlHZ24屬于HD-Zip I蛋白家族本文以維生素C合成的D-mannose/L-galactose途徑中關鍵GMP基因為切入點。GDP-D甘露糖焦磷酸化酶(GMP)在D-甘露糖/L半乳糖通路中扮演了重要的角色,據報道GMP家族中只有過表達SIGMP3提高了抗壞血酸的表達且改善了氧化應激耐受性。因此,本文使用酵母單雜交找尋與SIGMP3啟動子作用調節抗壞血酸的轉錄因子,共找到25個潛在蛋白,確認SlHZ24 為研究對象。序列分析顯示SlHZ24含有HD和LZ結構域(圖1a),且與其他HD-Zip I蛋白高度同源(圖1b)。進化分析的結果也說明SlHZ24與擬南芥的HD-Zip I蛋白同源。因此,SlHZ24屬于HD-Zip I蛋白。 2.酵母單/雙雜交驗證SlHZ24反式激活作用酵母雙雜交驗證了SlHZ24反式激活,含pBD-SlHZ24的實驗組可在SD/–Trp–His–Ade平板上生長,而對照組不生長(圖2a)。酵母單雜交驗證了不同長度SlHZ24和SIGMP3啟動子的作用,其中SlHZ24全長、N3、N4、N5、N6、N7包含HD結構域表現為正常生長,N1不包含HD結構域表現為細胞不生長,N2包含不完整的HD結構域表現為生長緩慢,說明HD結構域為互做結構域(圖2a)。
2.酵母單/雙雜交驗證SlHZ24反式激活作用酵母雙雜交驗證了SlHZ24反式激活,含pBD-SlHZ24的實驗組可在SD/–Trp–His–Ade平板上生長,而對照組不生長(圖2a)。酵母單雜交驗證了不同長度SlHZ24和SIGMP3啟動子的作用,其中SlHZ24全長、N3、N4、N5、N6、N7包含HD結構域表現為正常生長,N1不包含HD結構域表現為細胞不生長,N2包含不完整的HD結構域表現為生長緩慢,說明HD結構域為互做結構域(圖2a)。 3. 瞬轉及雙螢光素酶檢測驗證SlHZ24與SlGMP3啟動子結合將SlGMP3啟動子分成4段進行雙螢光素酶檢測實驗(圖3a、3b),P1和P2啟動子區域檢測到熒光增強,實驗結果說明SlHZ24結合SlGMP3啟動子的第二段序列,即-2411到-2313bp為結合區段。
3. 瞬轉及雙螢光素酶檢測驗證SlHZ24與SlGMP3啟動子結合將SlGMP3啟動子分成4段進行雙螢光素酶檢測實驗(圖3a、3b),P1和P2啟動子區域檢測到熒光增強,實驗結果說明SlHZ24結合SlGMP3啟動子的第二段序列,即-2411到-2313bp為結合區段。 4.進一步驗證SlHZ24和SlGMP3結合酵母單雜交驗證不同的SlGMP3啟動子和SlHZ24的結合情況(圖4a),結果顯示SlHZ24蛋白與SlGMP3啟動子的結合位點為-2411到-2313bp,突變實驗(圖4b)、EMSA實驗(圖4c)也證實了這點。這些結果表明SlHZ24直接與SlGMP3的上游元件結合來啟動下游表達。
4.進一步驗證SlHZ24和SlGMP3結合酵母單雜交驗證不同的SlGMP3啟動子和SlHZ24的結合情況(圖4a),結果顯示SlHZ24蛋白與SlGMP3啟動子的結合位點為-2411到-2313bp,突變實驗(圖4b)、EMSA實驗(圖4c)也證實了這點。這些結果表明SlHZ24直接與SlGMP3的上游元件結合來啟動下游表達。 5. 不同番茄組織及轉基因番茄中SlGMP3和SlHZ24檢測使用qPCR對番茄不同組織中的SlGMP3啟動子和SlHZ24的表達量進行了檢測,發現兩者都在莖和葉總的表達量較高(圖5a),說明SlHZ24正向調節SlGMP3表達。為了驗證SlHZ24對SlGMP3的影響,分別建立的SlHZ24過表達和干擾轉基因番茄進行下一步實驗(圖5b)。
5. 不同番茄組織及轉基因番茄中SlGMP3和SlHZ24檢測使用qPCR對番茄不同組織中的SlGMP3啟動子和SlHZ24的表達量進行了檢測,發現兩者都在莖和葉總的表達量較高(圖5a),說明SlHZ24正向調節SlGMP3表達。為了驗證SlHZ24對SlGMP3的影響,分別建立的SlHZ24過表達和干擾轉基因番茄進行下一步實驗(圖5b)。 6. SlHZ24通過調節SlGMP3表達來改變AsA的合成對過表達、干擾番茄葉及果實中AsA的檢測(圖6a 、6b)實驗,說明SlHZ24在番茄葉和果實中同時調節抗壞血酸的合成。過表達SlHZ24的OX-13植株果實中SlGMP3基因的表達量明顯高于野生型,而干擾SlHZ24的KD-9植株果實中SlGMP3基因的表達量變化不大(圖6d)。有趣的是在干擾SlHZ24的植株葉子中SlGMP3基因的表達量顯著減少,而過表達SlHZ24的植株葉子中SlGMP3基因的表達量變化不大(圖6c)。對轉基因番茄中其他GMP家族基因的檢測結果說明SlHZ24可能調節多種SIGMP基因(圖6c、圖6d)。
6. SlHZ24通過調節SlGMP3表達來改變AsA的合成對過表達、干擾番茄葉及果實中AsA的檢測(圖6a 、6b)實驗,說明SlHZ24在番茄葉和果實中同時調節抗壞血酸的合成。過表達SlHZ24的OX-13植株果實中SlGMP3基因的表達量明顯高于野生型,而干擾SlHZ24的KD-9植株果實中SlGMP3基因的表達量變化不大(圖6d)。有趣的是在干擾SlHZ24的植株葉子中SlGMP3基因的表達量顯著減少,而過表達SlHZ24的植株葉子中SlGMP3基因的表達量變化不大(圖6c)。對轉基因番茄中其他GMP家族基因的檢測結果說明SlHZ24可能調節多種SIGMP基因(圖6c、圖6d)。 7. 番茄中AsA和SlGMP3檢測為了檢測番茄果實成熟過程中SlHZ24如何影響SlGMP3,對不同時期果實的AsA和SlGMP3表達量進行了檢測(圖7a、7b),結果表明SlHZ24在番茄成熟的過程中可以調節SlGMP3的表達,其中以破色期果實調控效應最為顯著,除了紅果實成熟期。
7. 番茄中AsA和SlGMP3檢測為了檢測番茄果實成熟過程中SlHZ24如何影響SlGMP3,對不同時期果實的AsA和SlGMP3表達量進行了檢測(圖7a、7b),結果表明SlHZ24在番茄成熟的過程中可以調節SlGMP3的表達,其中以破色期果實調控效應最為顯著,除了紅果實成熟期。 8. SlHZ24轉基因番茄中D-甘露糖/L半乳糖通路其他基因的表達檢測使用qPCR對番茄的葉和果實進行了檢測(圖8a、8b),結果表明在野生型和轉基因番茄中SIGME2、SIGGP基因表達量發生了改變,顯示SlHZ24的多點協同調控作用。
8. SlHZ24轉基因番茄中D-甘露糖/L半乳糖通路其他基因的表達檢測使用qPCR對番茄的葉和果實進行了檢測(圖8a、8b),結果表明在野生型和轉基因番茄中SIGME2、SIGGP基因表達量發生了改變,顯示SlHZ24的多點協同調控作用。 9. SIGME2、SIGGP啟動子實驗使用EMSA實驗驗證轉錄因子SlHZ24和啟動子SIGME2、SIGGP有互做關系。
9. SIGME2、SIGGP啟動子實驗使用EMSA實驗驗證轉錄因子SlHZ24和啟動子SIGME2、SIGGP有互做關系。 10. SlHZ24光響應及氧化應激的作用為了研究光對AsA的作用,作者對不同光處理番茄中的AsA和SIHZ24的表達量進行了檢測(圖10a、10b),結果表明SIHZ24可以被光激活且AsA的表達也隨之提高。使用MV對轉基因番茄進行處理,同時對葉綠素和MDA濃度進行檢測,說明SIHZ24過表達可以提高植物對氧化應激的耐受性。
10. SlHZ24光響應及氧化應激的作用為了研究光對AsA的作用,作者對不同光處理番茄中的AsA和SIHZ24的表達量進行了檢測(圖10a、10b),結果表明SIHZ24可以被光激活且AsA的表達也隨之提高。使用MV對轉基因番茄進行處理,同時對葉綠素和MDA濃度進行檢測,說明SIHZ24過表達可以提高植物對氧化應激的耐受性。 文章小結目前植物合成AsA基礎途徑已經明確,但對AsA合成代謝的調控機制知之甚少。本文研究了番茄轉錄因子SlHZ24結合SIGMP3啟動子從而增加AsA表達水平的機制,進而提高植物對氧化應激的耐受性。AsA(又稱維C),是生物體一類小分子抗氧化劑物質,能清除體內活性氧,增強有機體抗氧化能力,保證新陳代謝正常進行。不過人類自身不能合成維C,必須從食物中攝取。本研究首次發現調控果實AsA積累的轉錄因子,對揭示AsA調控機制和未來品質改良具有重要意義。備注:本文出于傳遞更多信息為目的解讀該文獻內容,不希望轉載個人可與我聯系,會立即刪除處理。
文章小結目前植物合成AsA基礎途徑已經明確,但對AsA合成代謝的調控機制知之甚少。本文研究了番茄轉錄因子SlHZ24結合SIGMP3啟動子從而增加AsA表達水平的機制,進而提高植物對氧化應激的耐受性。AsA(又稱維C),是生物體一類小分子抗氧化劑物質,能清除體內活性氧,增強有機體抗氧化能力,保證新陳代謝正常進行。不過人類自身不能合成維C,必須從食物中攝取。本研究首次發現調控果實AsA積累的轉錄因子,對揭示AsA調控機制和未來品質改良具有重要意義。備注:本文出于傳遞更多信息為目的解讀該文獻內容,不希望轉載個人可與我聯系,會立即刪除處理。SWATH分析玉米蛋白質豐度、轉錄水平和組織多樣性揭示其發育調控規律
2018-04-27 17:25:38
文獻:An integrated analysis of protein abundance, transcript. level and tissue diversity to reveal developmental regulation of maize.Journal of Proteome Research(IF4.268)關鍵技術:SWATHSWATH分析玉米蛋白質豐度、轉錄水平和組織多樣性揭示發育調控規律研究背景采用SWATH-MS技術定量分析四種玉米組織中的蛋白質:未成熟雌穗(V7階段),未成熟雄穗(V8階段),授粉后20天(20_DAP)幼胚和14日齡幼苗根,并進行蛋白組&轉錄組關聯分析,鑒定組織特異性高表達基因和蛋白質,了解組織結構和器官發生的調節機制。研究內容1)數據分析和蛋白質特性合并來自四個組織樣本的蛋白組學數據,作者通過SWATH共獲得646,658個圖譜。用不同的置信度閾值和錯誤發現率(FDR)來評估SWATH-MS的檢測能力。發現。一定程度上較低閾值可鑒定到更多獨特肽和蛋白質(圖1A,1B)。在置信度0.85和FDR 0.05下,檢測的多肽和蛋白質數量與使用更嚴格的閾值標準—置信度0.90和FDR 0.02相比,檢測的多肽和蛋白數量稍有增加(圖1A,1B),但差異蛋白質數量變化不大(圖1D)。因此,最終選擇置信度0.85和FDR 0.05閾值,因為該閾值能檢測更多的蛋白質來擴增蛋白質組數據,而不會顯著增加差異蛋白數量。共有117,184個unique譜圖匹配到10,606個unique肽段,最后共鑒定到4,551個蛋白質(圖1E)。蛋白質豐度在重復實驗中重復性很好,相關系數在0.84-0.90之間(圖S1)。在雌穗,雄穗,幼胚和幼根中分別鑒定到3916、3707、3702和2871種蛋白質,在這四種組織中共鑒定到2269種蛋白質(圖1E)。大多數蛋白質(70%)被不少于2個unique譜圖覆蓋,主要由10至25個氨基酸組成(圖S2A,S2B)。大約64%的蛋白質顯示出> 5%的蛋白序列覆蓋率度,84%的蛋白質的分子質量> 20kDa。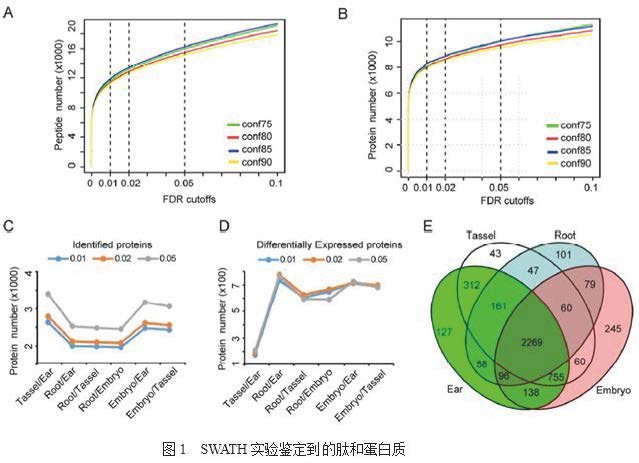 2)蛋白質組&轉錄組關聯分析基于Doreen Ware實驗室的轉錄組數據,及qTeller中下載的RNA-seq數據,作者對蛋白質組和轉錄組數據進行了關聯分析。先將每個組織中的蛋白質豐度與相應的mRNA水平進行比較。然后過濾掉那些低mRNA水平(RPKM<1)的蛋白質,總共保留了4,314種(94.8%)蛋白,其中雌穗3554個,雄穗3404個,幼胚3417個和幼根2370個(圖2A)。四個組織中共鑒定到2045個蛋白,表明大量檢測到的蛋白存在于各個組織中。此外,還鑒定到了組織特異性高度表達的蛋白,其中幼胚253個,幼根181個,雌穗123個和雄穗43個(圖2A)。其次,盡管蛋白質豐度與組織中的mRNA水平呈正相關,但Pearson相關系數很低,從0.35(幼根)到0.43(幼胚)(圖2B),這表明轉錄水平并不總是與蛋白質豐度一一對應。去掉組織特異性高度表達的蛋白質,以變化倍數> 2倍和P值<0.01,FDR <0.05為篩選條件,共有3714個差異蛋白至少在兩個組織中共同鑒定到。結果表明,在雌穗和雄穗之間鑒定到227個差異蛋白,在雌穗和幼根之間鑒定到799個差異蛋白(圖2D-E)。差異蛋白質的數量反映了各組織形態和功能的差異。為了探究蛋白質與轉錄本豐度相關性較低的原因,作者將這些差異蛋白質分成如下三個亞組:i)正相關亞組(PC):mRNA水平與蛋白質豐度正相關;ii)低相關性亞組(LC):編碼差異蛋白的轉錄本與蛋白質豐度相關但不顯著;iii)負相關亞組(NC):mRNA水平與蛋白質豐度呈負相關。作者發現大約50%的差異蛋白質可以歸類到PC亞組中(圖2E),這表明過半的差異蛋白質的差異豐度主要由這些基因的表達差異所決定。然而,大約42%的差異蛋白,從31%到62%不等,歸類到到LC亞組(圖2E)。這種現象通常是由于RNA轉錄,加工和轉換以及蛋白質翻譯和轉換的所導致的。4.9%至15%蛋白質歸類到NC亞組(圖2E),這表明轉錄組不能完全解釋在組織或器官的不同功能和形態的分子基礎上的蛋白質組,由此突出了蛋白質組學研究的重要性。
2)蛋白質組&轉錄組關聯分析基于Doreen Ware實驗室的轉錄組數據,及qTeller中下載的RNA-seq數據,作者對蛋白質組和轉錄組數據進行了關聯分析。先將每個組織中的蛋白質豐度與相應的mRNA水平進行比較。然后過濾掉那些低mRNA水平(RPKM<1)的蛋白質,總共保留了4,314種(94.8%)蛋白,其中雌穗3554個,雄穗3404個,幼胚3417個和幼根2370個(圖2A)。四個組織中共鑒定到2045個蛋白,表明大量檢測到的蛋白存在于各個組織中。此外,還鑒定到了組織特異性高度表達的蛋白,其中幼胚253個,幼根181個,雌穗123個和雄穗43個(圖2A)。其次,盡管蛋白質豐度與組織中的mRNA水平呈正相關,但Pearson相關系數很低,從0.35(幼根)到0.43(幼胚)(圖2B),這表明轉錄水平并不總是與蛋白質豐度一一對應。去掉組織特異性高度表達的蛋白質,以變化倍數> 2倍和P值<0.01,FDR <0.05為篩選條件,共有3714個差異蛋白至少在兩個組織中共同鑒定到。結果表明,在雌穗和雄穗之間鑒定到227個差異蛋白,在雌穗和幼根之間鑒定到799個差異蛋白(圖2D-E)。差異蛋白質的數量反映了各組織形態和功能的差異。為了探究蛋白質與轉錄本豐度相關性較低的原因,作者將這些差異蛋白質分成如下三個亞組:i)正相關亞組(PC):mRNA水平與蛋白質豐度正相關;ii)低相關性亞組(LC):編碼差異蛋白的轉錄本與蛋白質豐度相關但不顯著;iii)負相關亞組(NC):mRNA水平與蛋白質豐度呈負相關。作者發現大約50%的差異蛋白質可以歸類到PC亞組中(圖2E),這表明過半的差異蛋白質的差異豐度主要由這些基因的表達差異所決定。然而,大約42%的差異蛋白,從31%到62%不等,歸類到到LC亞組(圖2E)。這種現象通常是由于RNA轉錄,加工和轉換以及蛋白質翻譯和轉換的所導致的。4.9%至15%蛋白質歸類到NC亞組(圖2E),這表明轉錄組不能完全解釋在組織或器官的不同功能和形態的分子基礎上的蛋白質組,由此突出了蛋白質組學研究的重要性。 3)組織特異性高表達蛋白的功能分類基于蛋白質豐度作者在四種組織中共鑒定了600種組織特異性高表達蛋白質(圖3A)。在細胞的整個蛋白質組中,蛋白質的低覆蓋率是眾所周知的,在所有現有的蛋白質組技術,包括SWATH-MS都有明顯的缺陷。這種低覆蓋率可能導致對組織特異性高表達蛋白的數量的過高估計。因此,作者檢索了來自于qTeller的全長轉錄組數據和RNA-seq數據中編碼600個組織特異性高表達蛋白的基因的表達水平,發現大部分基因(84.3%)在多種組織中都有表達,只有94種蛋白編碼基因(15.7%)顯示出組織特異性表達模式(圖3A)。另外,26種蛋白質在雌穗和雄穗中共表達,稱為花序特異性蛋白質。作者利用qRT-PCR交叉驗證了18個基因的表達水平,15個為組織特異性表達基因,3個呈現出花序特異性表達模式(圖3B,3D)。此外,作者還利用Western blot驗證了2個幼胚特異性蛋白(GRMZM2G354013和GRMZM2G054916)(圖3C,3E)。作者鑒定了94個組織特異性蛋白,其中幼根60個,幼胚27個,雌穗5個和雄穗2個。幼根特異性蛋白質直接或間接地與氧化還原過程和脫氫有關,包括12個過氧化物酶超家族蛋白,3個還原酶和8個水解酶活性蛋白,表明對氧化脅迫的響應在幼根的發育和生長中起重要作用。在27個幼胚特異性蛋白中,有5種植物脂質轉移蛋白/種子儲存相關蛋白質,3種氧化還原酶活性蛋白質和2種已知的與光形態相關的蛋白質轉錄因子(AtPIF1和AtSPT轉錄因子)。7個花序特異性蛋白與花序結構和花器官的發育高度相關。值得注意的是,有18種蛋白質在雌穗和雄穗中都有很高的豐度,但其mRNA水平在四個組織中都呈現出的低水平或沒有被檢測到。
3)組織特異性高表達蛋白的功能分類基于蛋白質豐度作者在四種組織中共鑒定了600種組織特異性高表達蛋白質(圖3A)。在細胞的整個蛋白質組中,蛋白質的低覆蓋率是眾所周知的,在所有現有的蛋白質組技術,包括SWATH-MS都有明顯的缺陷。這種低覆蓋率可能導致對組織特異性高表達蛋白的數量的過高估計。因此,作者檢索了來自于qTeller的全長轉錄組數據和RNA-seq數據中編碼600個組織特異性高表達蛋白的基因的表達水平,發現大部分基因(84.3%)在多種組織中都有表達,只有94種蛋白編碼基因(15.7%)顯示出組織特異性表達模式(圖3A)。另外,26種蛋白質在雌穗和雄穗中共表達,稱為花序特異性蛋白質。作者利用qRT-PCR交叉驗證了18個基因的表達水平,15個為組織特異性表達基因,3個呈現出花序特異性表達模式(圖3B,3D)。此外,作者還利用Western blot驗證了2個幼胚特異性蛋白(GRMZM2G354013和GRMZM2G054916)(圖3C,3E)。作者鑒定了94個組織特異性蛋白,其中幼根60個,幼胚27個,雌穗5個和雄穗2個。幼根特異性蛋白質直接或間接地與氧化還原過程和脫氫有關,包括12個過氧化物酶超家族蛋白,3個還原酶和8個水解酶活性蛋白,表明對氧化脅迫的響應在幼根的發育和生長中起重要作用。在27個幼胚特異性蛋白中,有5種植物脂質轉移蛋白/種子儲存相關蛋白質,3種氧化還原酶活性蛋白質和2種已知的與光形態相關的蛋白質轉錄因子(AtPIF1和AtSPT轉錄因子)。7個花序特異性蛋白與花序結構和花器官的發育高度相關。值得注意的是,有18種蛋白質在雌穗和雄穗中都有很高的豐度,但其mRNA水平在四個組織中都呈現出的低水平或沒有被檢測到。 4)差異表達蛋白的功能分類GO富集確定差異蛋白功能類別。較其他組織對,雌穗和雄穗間鑒定到的差異蛋白更少,這些差異蛋白主要富集到“細胞分裂素介導的信號調節”,“乙酰輔酶A代謝過程”和“分生組織生長”(圖4A),表明細胞分裂素介導的信號在雌穗和雄穗間的細胞分裂活性存在差異。雄穗中UB3蛋白質豐度比雌穗中高1.7倍,表明UB蛋白質積累的增加可能支持雄穗長枝的長出。雌穗和幼胚間的差異蛋白主要富集到“脂質生物合成過程”,“分生組織起始”和“生殖結構發育”條目。雄穗和幼胚間的差異蛋白也富集到這些GO條目(圖4B,4C)。差異蛋白中不同GO類別反映了花序和幼胚間生理和發育上的差異。此外,雌穗/雄穗和幼根間的差異蛋白顯著富集到“對無機物質的響應”和“有機酸生物合成過程”(圖4D,4F)。幼胚和幼根間的差異蛋白主要富集到“有機酸代謝過程”,“對無機物質的反應”和“氧化還原”(p = 2.6 E-12)等。與“脂質生物合成過程”和“脂肪酸生物合成過程”有關的蛋白質在幼胚中顯著富集,而涉及“響應無機物質”,“氧化還原”和“醇代謝過程”的蛋白質在幼根中顯著富集。
4)差異表達蛋白的功能分類GO富集確定差異蛋白功能類別。較其他組織對,雌穗和雄穗間鑒定到的差異蛋白更少,這些差異蛋白主要富集到“細胞分裂素介導的信號調節”,“乙酰輔酶A代謝過程”和“分生組織生長”(圖4A),表明細胞分裂素介導的信號在雌穗和雄穗間的細胞分裂活性存在差異。雄穗中UB3蛋白質豐度比雌穗中高1.7倍,表明UB蛋白質積累的增加可能支持雄穗長枝的長出。雌穗和幼胚間的差異蛋白主要富集到“脂質生物合成過程”,“分生組織起始”和“生殖結構發育”條目。雄穗和幼胚間的差異蛋白也富集到這些GO條目(圖4B,4C)。差異蛋白中不同GO類別反映了花序和幼胚間生理和發育上的差異。此外,雌穗/雄穗和幼根間的差異蛋白顯著富集到“對無機物質的響應”和“有機酸生物合成過程”(圖4D,4F)。幼胚和幼根間的差異蛋白主要富集到“有機酸代謝過程”,“對無機物質的反應”和“氧化還原”(p = 2.6 E-12)等。與“脂質生物合成過程”和“脂肪酸生物合成過程”有關的蛋白質在幼胚中顯著富集,而涉及“響應無機物質”,“氧化還原”和“醇代謝過程”的蛋白質在幼根中顯著富集。 文章小結作者利用SWATH-MS技術在玉米4種組織中共鑒定到4551種蛋白質,與之前iTRAQ鑒定到的蛋白數目相當。基于蛋白質組和轉錄組關聯分析,作者推斷出蛋白質豐度與mRNA水平呈正相關,具有弱到中等的相關系數。然而,一些特定組織中與功能或結構相關的一些關鍵蛋白質在轉錄水平上被時空調控,在4種組織中不同蛋白表現出不同的表達模式。此外,組織特異性高表達的蛋白和差異蛋白能富集到不同GO類別中。組織特異性高度表達的蛋白質亞組通過交叉實驗驗證可作為潛在生物標志物。這加深了對玉米組織和器官發育分子機制的理解,并為其生物標志物的發現提供了全面的線索。備注:本文出于傳遞更多信息為目的解讀該文獻內容,不希望轉載個人可與我聯系,會立即刪除處理。
文章小結作者利用SWATH-MS技術在玉米4種組織中共鑒定到4551種蛋白質,與之前iTRAQ鑒定到的蛋白數目相當。基于蛋白質組和轉錄組關聯分析,作者推斷出蛋白質豐度與mRNA水平呈正相關,具有弱到中等的相關系數。然而,一些特定組織中與功能或結構相關的一些關鍵蛋白質在轉錄水平上被時空調控,在4種組織中不同蛋白表現出不同的表達模式。此外,組織特異性高表達的蛋白和差異蛋白能富集到不同GO類別中。組織特異性高度表達的蛋白質亞組通過交叉實驗驗證可作為潛在生物標志物。這加深了對玉米組織和器官發育分子機制的理解,并為其生物標志物的發現提供了全面的線索。備注:本文出于傳遞更多信息為目的解讀該文獻內容,不希望轉載個人可與我聯系,會立即刪除處理。lncRNAH19作為ceRNA通過調控RNAmiR-29b-3p來調節膀胱癌細胞上皮間質轉化和轉移
2017-12-28 11:44:48
文獻:lncRNA H19 regulates epithelial–mesenchymal transition and metastasis of bladder cancer by miR-29b-3p as competing endogenous RNA. Biochim Biophys Acta(IF6.261)關鍵技術:1、病毒轉染(慢病毒包裝、腺病毒包裝)2、分子互作技術(Pull down、RIP(MS2/Ago)、雙熒光素酶實驗)3、檢測分析(免疫組化、克隆形成、細胞劃痕愈合、CCK8、EdU、HE染色、WB、ELISA、q-PCR)4、分子定位技術(FISH、IHC、免疫熒光、激光共聚焦)5、芯片技術(lncRNA/mRNA芯片)長鏈非編碼RNAH19作為內源競爭性RNA通過調控RNAmiR-29b-3p來調節膀胱癌細胞上皮間質轉化和轉移
研究背景EMT(epithelial-to-mesenchymal transition)主要在腫瘤侵襲和轉移中發揮作用,其改變主要表現在分子、細胞、組織水平,涉及組織細胞形態改變、組織特異分子表達的改變。通過檢測相關分子表達水平或細胞形態改變可以評估腫瘤進展、了解藥物療效及基因功能作用。lncRNA(long noncoding RNAs)與腫瘤的發生發展關系密切,越來越多的研究表明,lncRNA可以調控腫瘤細胞的增殖、遷移、凋亡、分化進而調控腫瘤的發生發展。本文以膀胱癌和腫瘤侵襲為研究對象,聚焦EMT過程和lncRNA功能機制,以lncRNA調控EMT機制為主線,綜合運用芯片、生物信息學分析、分子互作技術(RIP、RNA pulldown、雙熒光素酶)、分子定位(免疫熒光、FISH)等技術研究了lncRNA的下游互作分子,探討了H19(lncRNA)功能機制。構建了H19調控膀胱癌侵襲、轉移的作用軸線(如下圖)。 研究內容及結果1、臨床研究水平,研究了正常人和病人H19和DNMT3B的表達水平和相關性。2、體外實驗,尋找并綜合運用多種技術手段驗證了H19的相互作用靶分子miR-29b-3p及miR-29b-3p的相互作用靶分子DNMT3B3、體內實驗,運用裸鼠成瘤實驗驗證了H19對腫瘤侵襲轉移的作用。一、臨床研究水平研究了癌組織和癌旁組織以及膀胱癌細胞系中H19和DNMT3B的表達水平和相關性。發現H19和DNMT3B在癌組織中高表達而在癌旁組織中低表達。運用pearson correlation分析發現H19和DNMT3B的高表達具有正相關性。這暗示了H19對DNMT3B可能具有正調控關系。這是后面作者進行生物信息學預測和機制功能研究的基礎。1、作者首先統計了臨床上H19的表達和膀胱癌患者性別、年齡、腫瘤病理分期、淋巴轉移的關系。發現高表達的H19和膀胱癌的分期和淋巴轉移存在密切關系(P<0.05)。
研究內容及結果1、臨床研究水平,研究了正常人和病人H19和DNMT3B的表達水平和相關性。2、體外實驗,尋找并綜合運用多種技術手段驗證了H19的相互作用靶分子miR-29b-3p及miR-29b-3p的相互作用靶分子DNMT3B3、體內實驗,運用裸鼠成瘤實驗驗證了H19對腫瘤侵襲轉移的作用。一、臨床研究水平研究了癌組織和癌旁組織以及膀胱癌細胞系中H19和DNMT3B的表達水平和相關性。發現H19和DNMT3B在癌組織中高表達而在癌旁組織中低表達。運用pearson correlation分析發現H19和DNMT3B的高表達具有正相關性。這暗示了H19對DNMT3B可能具有正調控關系。這是后面作者進行生物信息學預測和機制功能研究的基礎。1、作者首先統計了臨床上H19的表達和膀胱癌患者性別、年齡、腫瘤病理分期、淋巴轉移的關系。發現高表達的H19和膀胱癌的分期和淋巴轉移存在密切關系(P<0.05)。 2、運用芯片和q-PCR研究了H19和DNMT3B在膀胱癌和癌旁組織中的表達水平。芯片技術發現H19和DNMT3B在腫瘤組織中均高表達,而在癌旁組織中低表達,芯片的結果得到了q-PCR的驗證。
2、運用芯片和q-PCR研究了H19和DNMT3B在膀胱癌和癌旁組織中的表達水平。芯片技術發現H19和DNMT3B在腫瘤組織中均高表達,而在癌旁組織中低表達,芯片的結果得到了q-PCR的驗證。 3、Pearson correlation分析發現H19和DNMT3B的高表達具有正相關性,說明H19對DNMT3B的表達可能具有正調控作用,如果H19作為ceRNA,則可能存在miRNA來受lncRNA的調節來調控DNMT3B。
3、Pearson correlation分析發現H19和DNMT3B的高表達具有正相關性,說明H19對DNMT3B的表達可能具有正調控作用,如果H19作為ceRNA,則可能存在miRNA來受lncRNA的調節來調控DNMT3B。 4、在組織水平之外,作者也研究了常用的幾種膀胱癌細胞系中的H19表達水平,發現在常用的幾種癌細胞系中H19均高表達。
4、在組織水平之外,作者也研究了常用的幾種膀胱癌細胞系中的H19表達水平,發現在常用的幾種癌細胞系中H19均高表達。 二、體外實驗臨床實驗證實了H19與膀胱癌的分期、轉移有密切的關系,同時H19和DNMT3B可能存在正調控關系。作者綜合運用多種功能實驗研究了H19的體外功能,在此基礎上然后運用生物信息學分析方法和多種互作技術預測并且驗證了H19的互作分子miR-29b-3p和下游調控靶分子DNMT3B。1、功能研究。作者綜合運用CCK8、EdU、克隆形成、劃痕愈合、Transwell、免疫熒光等技術對H19的生物學功能進行了研究,發現H19可以促進癌細胞的增殖、侵襲、遷移、轉移和細胞骨架重構。
二、體外實驗臨床實驗證實了H19與膀胱癌的分期、轉移有密切的關系,同時H19和DNMT3B可能存在正調控關系。作者綜合運用多種功能實驗研究了H19的體外功能,在此基礎上然后運用生物信息學分析方法和多種互作技術預測并且驗證了H19的互作分子miR-29b-3p和下游調控靶分子DNMT3B。1、功能研究。作者綜合運用CCK8、EdU、克隆形成、劃痕愈合、Transwell、免疫熒光等技術對H19的生物學功能進行了研究,發現H19可以促進癌細胞的增殖、侵襲、遷移、轉移和細胞骨架重構。
 2、機制研究。生物學預測發現H19和DNMT3B都可以與miR-29b-3p結合。雙熒光素酶證實H19、DNMT3B可以與miR-29b-3p直接結合。FISH實驗可以發現H19和miR-29b-3p在細胞內存在共定位。GFP-MS2-RIP和RNA-pulldown實驗驗證了H19可以內源性結合miR-29b-3p。Anti-AGO2 RIP進一步驗證了H19可以通過AGO2與miR-29b-3p結合。而H19、DNMT3B和miR-29b-3p的三因子雙熒光素酶實驗最終確認三者的ceRNA調控網絡。
2、機制研究。生物學預測發現H19和DNMT3B都可以與miR-29b-3p結合。雙熒光素酶證實H19、DNMT3B可以與miR-29b-3p直接結合。FISH實驗可以發現H19和miR-29b-3p在細胞內存在共定位。GFP-MS2-RIP和RNA-pulldown實驗驗證了H19可以內源性結合miR-29b-3p。Anti-AGO2 RIP進一步驗證了H19可以通過AGO2與miR-29b-3p結合。而H19、DNMT3B和miR-29b-3p的三因子雙熒光素酶實驗最終確認三者的ceRNA調控網絡。









 三、體內實驗在幾個膀胱癌細胞系的體外機制研究表明,H19與miR-29b、miR-29b與DNMT3B之間存在調控關系,H19作為ceRNA可以通過直接調控miR-29b來調控DNMT3B的表達,進而調控下游相關分子的表達和EMT相關功能的實現。接著作者用裸鼠成瘤實驗驗證了H19、miR-29b對膀胱癌細胞成瘤能力、轉移能力、血管形成的影響。
三、體內實驗在幾個膀胱癌細胞系的體外機制研究表明,H19與miR-29b、miR-29b與DNMT3B之間存在調控關系,H19作為ceRNA可以通過直接調控miR-29b來調控DNMT3B的表達,進而調控下游相關分子的表達和EMT相關功能的實現。接著作者用裸鼠成瘤實驗驗證了H19、miR-29b對膀胱癌細胞成瘤能力、轉移能力、血管形成的影響。


 文章總結作者從臨床案例入手發現了膀胱癌與H19表達關系,然后運用芯片技術和q-PCR進一步驗證了膀胱癌中H19和DNMT3B存在正相關關系,生物信息學分析預測了由miR-29b調控DNMT3B表達的ceRNA網絡關系。體外實驗中,互作分子技術(雙熒光素酶實驗、RIP、RNA pulldown)驗證了生物信息學的預測,功能實驗驗證了H19、miR-29b和DNMT3B三種在細胞增殖、侵襲、遷移、EMT轉化中的作用和相互作用關系。接著作者進一步開展了體內實驗,裸鼠成瘤實驗證實了H19可以促進膀胱癌細胞的增殖、淋巴轉移、血管生成。這樣作者從臨床、體外、體內三個水平探究了作為ceRNA的lncRNA(H19)在調控膀胱癌細胞侵襲轉移EMT轉化中的調控網絡,描繪了H19/miR-29b/DNMT3B調控主線。
文章總結作者從臨床案例入手發現了膀胱癌與H19表達關系,然后運用芯片技術和q-PCR進一步驗證了膀胱癌中H19和DNMT3B存在正相關關系,生物信息學分析預測了由miR-29b調控DNMT3B表達的ceRNA網絡關系。體外實驗中,互作分子技術(雙熒光素酶實驗、RIP、RNA pulldown)驗證了生物信息學的預測,功能實驗驗證了H19、miR-29b和DNMT3B三種在細胞增殖、侵襲、遷移、EMT轉化中的作用和相互作用關系。接著作者進一步開展了體內實驗,裸鼠成瘤實驗證實了H19可以促進膀胱癌細胞的增殖、淋巴轉移、血管生成。這樣作者從臨床、體外、體內三個水平探究了作為ceRNA的lncRNA(H19)在調控膀胱癌細胞侵襲轉移EMT轉化中的調控網絡,描繪了H19/miR-29b/DNMT3B調控主線。本文采取病毒包裝、分子互作(Pull down、RIP、雙熒光素酶實驗)、檢測分析(免疫組化、WB、q-PCR)、分子定位(FISH、IHC、IF)、lncRNA/mRNA芯片等方法,研究lncRNAH19通過調控miR-29b-3p調節膀胱癌細胞上皮間質轉化和轉移機制。文獻全文索取、技術服務溝通可隨時@qq3268908524......
備注:本文章出于傳遞更多信息為目的解讀了該文獻部分內容,不希望轉載的個人可與我聯系,會立即刪除處理。
假單胞菌轉錄調節因子AlgR通過非編碼RNA RsmZ調控脂肪酶LipA表達探討
2017-12-25 14:34:47
參考文獻:The Pseudomonas transcriptional regulator AlgR controls LipA expression via the noncoding RNA RsmZ in Pseudomonas protegens Pf-5. Biochemical and Biophysical Research Communications(IF2.466)關鍵技術:EMSA、基因敲除CRISPR-Cas9、lacZ報告基因假單胞菌轉錄調節因子AlgR通過非編碼RNA RsmZ調控脂肪酶LipA表達機制探討
脂肪酶(Lipase)是一種存在于動物、植物、微生物中的工業用酶,廣泛應用于食品、去垢劑、生物質能等領域。目前商品化的脂肪酶主要來源于微生物,但傳統的育種、培養基營養優化的手段在提高其產量方面面臨瓶頸,亟待從分子機制層面尋找解決途徑。目前研究證實細菌中脂肪酶的表達受到雙組份調控體系如LipQ-LipR調節,但假單胞菌P. protegens Pf-5菌株的分子調控機制有待深入研究,AlgR作為轉錄調控因子在假單胞菌P. protegens Pf-5菌株中對于脂肪酶產量控制關系及機理此前還未見報道,同時已知AlgR也能夠調節ncRNA RsmZ,故而本文采取基因敲除、lacZ報告基因、EMSA等方法研究AlgR對于lipA的表達調控機制。研究內容及結果1. 證實AlgR在轉錄水平控制lipA表達構建了algR敲除的Pf-5菌株(名為Pf6003),培養測定其生長曲線,并檢測對比敲除菌株中lipA酶活性。結果顯示野生型、algR敲除型Pf-5菌株的呈現一致生長曲線,說明algR的敲除不影響Pf-5的生長(圖A),但是敲除株的總脂肪酶活性顯著下降(圖B)。 此外將lipA的啟動子連接lacZ報告基因、將報告基因質粒分別轉入野生型及突變型Pf-5菌株,后續檢測β半乳糖苷酶(b-galactosidase)活性,發現在敲除突變株中b半乳糖苷酶活性低于野生型(圖C)。通過RT-PCR檢測lipA的mRNA表達,顯示突變株中lipA mRNA水平降低(圖D)。
此外將lipA的啟動子連接lacZ報告基因、將報告基因質粒分別轉入野生型及突變型Pf-5菌株,后續檢測β半乳糖苷酶(b-galactosidase)活性,發現在敲除突變株中b半乳糖苷酶活性低于野生型(圖C)。通過RT-PCR檢測lipA的mRNA表達,顯示突變株中lipA mRNA水平降低(圖D)。 同時通過lacZ報告基因實驗顯示algR的補償過表達在野生型和缺失突變型Pf-5中都能提高β半乳糖苷酶活性及lipA酶活性。
同時通過lacZ報告基因實驗顯示algR的補償過表達在野生型和缺失突變型Pf-5中都能提高β半乳糖苷酶活性及lipA酶活性。 2. AlgR影響rsmX/rsmY/rsmZ的表達使用RT-PCR實驗證實在Pf-5菌株中敲除AlgR基因后,rsmX/rsmY/rsmZ的轉錄水平均上調。
2. AlgR影響rsmX/rsmY/rsmZ的表達使用RT-PCR實驗證實在Pf-5菌株中敲除AlgR基因后,rsmX/rsmY/rsmZ的轉錄水平均上調。 3. AlgR直接結合于rsmZ啟動子序列本文表達純化了AlgR蛋白、合成了rsmX/rsmY/rsmZ啟動子序列的探針,進行EMSA實驗驗證AlgR蛋白同這些基因啟動子序列的互作關系,證實AlgR蛋白存在直接相互作用、而AlgR蛋白不能結合于rsmX/rsmY的啟動子序列。
3. AlgR直接結合于rsmZ啟動子序列本文表達純化了AlgR蛋白、合成了rsmX/rsmY/rsmZ啟動子序列的探針,進行EMSA實驗驗證AlgR蛋白同這些基因啟動子序列的互作關系,證實AlgR蛋白存在直接相互作用、而AlgR蛋白不能結合于rsmX/rsmY的啟動子序列。 5. AlgR通過rsmZ調節lipA的表達構建了rsmZ敲除的Pf-5菌株(名為Pf6285),并構建了同時雙敲除algR和rsmZ的Pf-5菌株(名為Pf6100),通過lazZ報告基因及酶活力測定顯示,同時敲除algR和rsmZ基因的Pf6100中lipA表達及總酶活性顯著低于algR單獨敲除的菌株,而在algR單獨敲除株中過表達rsmZ能夠使lipA酶表達及活性恢復到野生型水平。
5. AlgR通過rsmZ調節lipA的表達構建了rsmZ敲除的Pf-5菌株(名為Pf6285),并構建了同時雙敲除algR和rsmZ的Pf-5菌株(名為Pf6100),通過lazZ報告基因及酶活力測定顯示,同時敲除algR和rsmZ基因的Pf6100中lipA表達及總酶活性顯著低于algR單獨敲除的菌株,而在algR單獨敲除株中過表達rsmZ能夠使lipA酶表達及活性恢復到野生型水平。 總結構建了分別單獨敲除algR或rsmZ的假單胞菌株Pf-5,及同時敲除algR和rsmZ的菌株,通過lacZ報告基因系統以及細胞總酶活力測定,證實了algR的缺失突變導致lipA水平下降,同時rsmZ的過表達能夠補償algR缺失對于lipA的下調效應,并通過EMSA實驗驗證了AlgR蛋白能直接結合于rsmZ的啟動子序列,說明algR基因是通過rsmZ來調控假單胞菌株Pf-5中lipA的表達水平。
總結構建了分別單獨敲除algR或rsmZ的假單胞菌株Pf-5,及同時敲除algR和rsmZ的菌株,通過lacZ報告基因系統以及細胞總酶活力測定,證實了algR的缺失突變導致lipA水平下降,同時rsmZ的過表達能夠補償algR缺失對于lipA的下調效應,并通過EMSA實驗驗證了AlgR蛋白能直接結合于rsmZ的啟動子序列,說明algR基因是通過rsmZ來調控假單胞菌株Pf-5中lipA的表達水平。miR-124a通過沉默FOXA2導致細胞內脂質積聚異常的機制探討
2017-12-25 13:46:00
參考文獻:Mesenchyma stem cells as natural bio-factories for exosomes carrying miR-124a in the treatment of gliomas. Neuro-Oncology(IF7.786)關鍵技術:1. 重組病毒包裝:慢病毒包裝、穩轉株;2. 分子生物學:Cre-loxP位點特異性重組酶系統;3. 檢測分析:WST-1、Transwell、qRT-PCR、Western blot、納米顆粒跟蹤分析技術;4. 動物實驗:裸鼠顱內移植瘤模型miR-124a通過沉默FOXA2導致細胞內脂質積聚異常從而引起細胞凋亡
膠質母細胞瘤是星形細胞腫瘤中惡性程度最高的膠質瘤,已有研究表明microRNAs(miRs)有希望成為膠質母細胞瘤的新型治療方法。然而,何種miRs對抗膠質母細胞瘤的效果最佳,以及如何將miRs遞送至膠質母細胞瘤是目前尚未解決的問題。本文通過功能篩選,作者發現miR-124a抗膠質瘤的效果最為顯著,使用含有miR-124a的慢病毒載體感染間充質干細胞(MSCs),并用分離獲得的外泌體處理膠質瘤干細胞(GSCs)后發現GSCs活力顯著下降,體內實驗證實Exo-miR124能使顱內移植GSCs的小鼠存活更久。研究表明miR-124a通過沉默FOXA2導致細胞內脂質積聚異常從而引起細胞凋亡。因此,MSCs可以作為一個產生exo-miR124的天然生物工廠來參與抗膠質瘤治療。研究內容及結果1. miR-124a是一種能有效抗膠質瘤的microRNA基于數據查詢,作者選取8個研究報道具有抗膠質瘤特性的miRs進行初步篩選。采用慢病毒載體將這些miRs分別轉移至5種不同的GSCs,通過WST-1測定發現在所有GSCs細胞系中miR-124a引起細胞增殖能力降低的效果最顯著,表明miR-124是最為有效的抗膠質瘤microRNA。 2.MSCs能夠有效地將miR-124a轉移至GSCs作者使用慢病毒包裝miR-124a并感染MSCs,通過Transwell與WST-1以及臺盼藍染色結合證實MSCs-miR124導致GSCs的增值能力顯著下降,暗示MSCs可通過某種方式將miR-124a轉移至GSCs。
2.MSCs能夠有效地將miR-124a轉移至GSCs作者使用慢病毒包裝miR-124a并感染MSCs,通過Transwell與WST-1以及臺盼藍染色結合證實MSCs-miR124導致GSCs的增值能力顯著下降,暗示MSCs可通過某種方式將miR-124a轉移至GSCs。 3. MSCs將miR-124a包裝至外泌體并進一步轉移至GSCs作者通過透射電鏡觀察、納米顆粒跟蹤分析技術結合對外泌體標記蛋白的Western blot檢測證實MSCs釋放的顆粒是外泌體,且外泌體中miR-124a的含量顯著上調。進一步通過Cre-loxP位點特異性重組酶系統結合qRT-PCR和Western blot檢測證實外泌體能夠將功能性的RNA分子轉移至腫瘤細胞。
3. MSCs將miR-124a包裝至外泌體并進一步轉移至GSCs作者通過透射電鏡觀察、納米顆粒跟蹤分析技術結合對外泌體標記蛋白的Western blot檢測證實MSCs釋放的顆粒是外泌體,且外泌體中miR-124a的含量顯著上調。進一步通過Cre-loxP位點特異性重組酶系統結合qRT-PCR和Western blot檢測證實外泌體能夠將功能性的RNA分子轉移至腫瘤細胞。 4. 攜帶miR-124a的外泌體在小鼠體內能夠有效對抗膠質瘤研究表明,在裸鼠額葉內移植GSC267細胞一周后,分別通過腹腔注射和靜脈注射攜帶miR124的外泌體(Exo-miR124)能夠使裸鼠存活時間更長。組織切片結果表明,裸鼠腦部的腫瘤完全消退,證實Exo-miR124在小鼠體內同樣能夠有效對抗膠質瘤。
4. 攜帶miR-124a的外泌體在小鼠體內能夠有效對抗膠質瘤研究表明,在裸鼠額葉內移植GSC267細胞一周后,分別通過腹腔注射和靜脈注射攜帶miR124的外泌體(Exo-miR124)能夠使裸鼠存活時間更長。組織切片結果表明,裸鼠腦部的腫瘤完全消退,證實Exo-miR124在小鼠體內同樣能夠有效對抗膠質瘤。 5. Exo-miR124抑制FOXA2的表達并改變GSCs的脂質代謝作者通過體內和體外實驗檢測已知的miR124-a靶基因蛋白表達水平,發現僅FOXA2的表達量顯著下調。慢病毒介導的不同水平的FOXA2干擾表達同樣引起GSCs的增殖能力,從而產生對應劑量效應的下降。互補實驗以及進一步的油紅O染色和Western blot檢測結果表明,miR-124a通過下調FOXA2的表達引起細胞內脂質積累,最終導致腫瘤細胞凋亡。
5. Exo-miR124抑制FOXA2的表達并改變GSCs的脂質代謝作者通過體內和體外實驗檢測已知的miR124-a靶基因蛋白表達水平,發現僅FOXA2的表達量顯著下調。慢病毒介導的不同水平的FOXA2干擾表達同樣引起GSCs的增殖能力,從而產生對應劑量效應的下降。互補實驗以及進一步的油紅O染色和Western blot檢測結果表明,miR-124a通過下調FOXA2的表達引起細胞內脂質積累,最終導致腫瘤細胞凋亡。 總結首先通過篩選確定了miR-124a是最有效的抗膠質瘤microRNA,以慢病毒為載體將miR-124a轉移至MSCs后發現其分泌的外泌體中同樣攜帶miR-124a,體內體外實驗均表明Exo-miR124能夠抑制GSCs的增殖。進一步的機理研究發現miR-124a通過下調FOXA2的表達從而引起細胞內脂質積累,導致脂質代謝異常并最終引起腫瘤細胞凋亡。本文首次提出MSCs可以用于離體生產攜帶抗膠質瘤microRNA的外泌體,以期實現消除惡性神經膠質瘤。以上是miR-124a通過沉默FOXA2導致細胞內脂質積聚異常從而引起細胞凋亡的探討內容及結果,供學習參考!@3268908524索取文獻全文,咨詢服務外包均可。miRNA靶基因篩選及驗證 非編碼RNA研究.外泌體研究.表觀遺傳調控......
總結首先通過篩選確定了miR-124a是最有效的抗膠質瘤microRNA,以慢病毒為載體將miR-124a轉移至MSCs后發現其分泌的外泌體中同樣攜帶miR-124a,體內體外實驗均表明Exo-miR124能夠抑制GSCs的增殖。進一步的機理研究發現miR-124a通過下調FOXA2的表達從而引起細胞內脂質積累,導致脂質代謝異常并最終引起腫瘤細胞凋亡。本文首次提出MSCs可以用于離體生產攜帶抗膠質瘤microRNA的外泌體,以期實現消除惡性神經膠質瘤。以上是miR-124a通過沉默FOXA2導致細胞內脂質積聚異常從而引起細胞凋亡的探討內容及結果,供學習參考!@3268908524索取文獻全文,咨詢服務外包均可。miRNA靶基因篩選及驗證 非編碼RNA研究.外泌體研究.表觀遺傳調控......iTRAQ蛋白組學技術應用
2017-12-01 13:42:48
iTRAQ(isobaric tags for relative and absolute quantitation)技術是由AB SCIEX公司研發的一種體外同重同位素標記的相對與絕對定量技術,利用多種同位素試劑標記蛋白多肽N末端或賴氨酸側鏈基團,經高精度質譜儀串聯分析,可同時比較多達8種樣品之間的蛋白表達量,是近年來定量蛋白質組學中應用最廣泛的高通量篩選技術。那么iTRAQ試劑主要構成呢?iTRAQ試劑分為報告基團、平衡基團、氨基酸反應基團,報告基團共有8種(113、114、115、116、117、118、119、121Da),與平衡基團質量相加正好是305Da。氨基酸反應基團與肽鏈N端賴氨酸側鏈共價鏈接,從而將報告基團和平衡基團標記到肽段上。在一級質譜中,不同來源的相同肽段被連接上總質量相同的iTRAQ標簽試劑,具有相同的質荷比,表現為一個峰。在二級質譜中,iTRAQ標簽試劑在不同基團連接處發生斷裂,表現出不同質荷比的峰(平衡基團發生中性丟失)。根據波峰高度和面積,可以得到同一蛋白在不同樣本中或在不同處理條件下的表達差異。 iTRAQ技術作為經典蛋白質組學定量技術,它的優勢?靈敏度高:可檢測出較低豐度蛋白,胞漿蛋白、膜蛋白、核蛋白、胞外蛋白等;分離能力強:可分離出酸/堿性蛋白,小于 10K 或大于200K的蛋白、難溶性蛋白;高通量:可同時對8個樣本進行分析,特別適用于采用多種處理方式或來自多個處理時間的樣本的差異蛋白分析;結果可靠準確:定性與定量同步進行,同時得出鑒定和定量結果,重復樣品間的蛋白表達量相關性可達到 0.95 以上;自動化程度高:液質連用,自動化操作,分析速度快,分離效果好。除了涉及iTRAQ蛋白質組學實驗樣品、實驗流程、質譜下機原始數據等,質譜數據分析也顯得尤為重要,試想拿到數據卻無法分析,無法得知實驗最終結果是否與預期相符,可謂是萬里長征只差一步。大數據時代的到來引起了高校對大數據分析關注,很多都陸續添加了生物信息學、生物統計學等課程,如果此刻的您也很感興趣,不妨多深究下。iTRAQ蛋白質組學--生物信息學分析內容:iTRAQ鑒定:包括鑒定結果統計、重復性分析、Unique肽段數分布、肽段長度分布、蛋白覆蓋度分布;iTRAQ定量:包括定量信息統計、蛋白質豐度比(FC)分布、表達量層次聚類分析;蛋白質功能注釋:包括GO注釋、COG注釋和Pathway代謝通路注釋;差異蛋白的富集分析:包括差異蛋白的GO富集分析、差異蛋白的Pathway富集分析。如果要做iTRAQ蛋白質組服務,需要注意什么呢?樣本,外包公司銷售會和您溝通的,不同樣本由于高豐度/低豐度蛋白含量不同也會有差異的,還不如先讓對方技術評估下實驗方案。可能要強調一點,最后質譜下機原始數據需要結合數據庫檢索,所以要求物種具有蛋白質參考數據庫、EST 序列(轉錄組)或基因組注釋信息等,不清楚的,可以請外包公司幫您查下。iTRAQ現今發表文獻網上有很多,可以下載學習,通過文獻可以更清楚了解iTRAQ技術應用,也可@3268908524索取相關iTRAQ發表文獻,加深對iTRAQ蛋白質組學的理解,拓寬項目實驗設計思路。下期接著回歸到非編碼RNA上,也是小編對非編碼RNA很感興趣,就是這么任性!換個方向,也換個視野,知識都需要消化吸收,才能更好地加以利用。一本正經地說,噗嗤……
iTRAQ技術作為經典蛋白質組學定量技術,它的優勢?靈敏度高:可檢測出較低豐度蛋白,胞漿蛋白、膜蛋白、核蛋白、胞外蛋白等;分離能力強:可分離出酸/堿性蛋白,小于 10K 或大于200K的蛋白、難溶性蛋白;高通量:可同時對8個樣本進行分析,特別適用于采用多種處理方式或來自多個處理時間的樣本的差異蛋白分析;結果可靠準確:定性與定量同步進行,同時得出鑒定和定量結果,重復樣品間的蛋白表達量相關性可達到 0.95 以上;自動化程度高:液質連用,自動化操作,分析速度快,分離效果好。除了涉及iTRAQ蛋白質組學實驗樣品、實驗流程、質譜下機原始數據等,質譜數據分析也顯得尤為重要,試想拿到數據卻無法分析,無法得知實驗最終結果是否與預期相符,可謂是萬里長征只差一步。大數據時代的到來引起了高校對大數據分析關注,很多都陸續添加了生物信息學、生物統計學等課程,如果此刻的您也很感興趣,不妨多深究下。iTRAQ蛋白質組學--生物信息學分析內容:iTRAQ鑒定:包括鑒定結果統計、重復性分析、Unique肽段數分布、肽段長度分布、蛋白覆蓋度分布;iTRAQ定量:包括定量信息統計、蛋白質豐度比(FC)分布、表達量層次聚類分析;蛋白質功能注釋:包括GO注釋、COG注釋和Pathway代謝通路注釋;差異蛋白的富集分析:包括差異蛋白的GO富集分析、差異蛋白的Pathway富集分析。如果要做iTRAQ蛋白質組服務,需要注意什么呢?樣本,外包公司銷售會和您溝通的,不同樣本由于高豐度/低豐度蛋白含量不同也會有差異的,還不如先讓對方技術評估下實驗方案。可能要強調一點,最后質譜下機原始數據需要結合數據庫檢索,所以要求物種具有蛋白質參考數據庫、EST 序列(轉錄組)或基因組注釋信息等,不清楚的,可以請外包公司幫您查下。iTRAQ現今發表文獻網上有很多,可以下載學習,通過文獻可以更清楚了解iTRAQ技術應用,也可@3268908524索取相關iTRAQ發表文獻,加深對iTRAQ蛋白質組學的理解,拓寬項目實驗設計思路。下期接著回歸到非編碼RNA上,也是小編對非編碼RNA很感興趣,就是這么任性!換個方向,也換個視野,知識都需要消化吸收,才能更好地加以利用。一本正經地說,噗嗤……蛋白質-核酸相互作用研究方法介紹
2017-11-23 16:35:13
蛋白質和核酸是組成生命的主要生物大分子,研究蛋白質-核酸相互作用、蛋白質-蛋白質相互作用是后基因組時代重要的研究領域之一。專注蛋白相關研究, 金開瑞可提供凝膠電泳遷移率EMSA、染色質免疫沉淀ChIP、RNA Pull Down / DNA Pull Down、熒光素酶報告基因、酵母單雜交等多種蛋白質-核酸相互作用研究服務,幫助您理清轉錄調控過程,預測互作蛋白,加速研究進程。1) 凝膠電泳遷移率EMSA凝膠遷移或電泳遷移率檢測(Electrophoretic Mobility Shift Assay,EMSA)是一種檢測蛋白質和DNA序列相互結合的技術,可用于定性和定量分析;目前已用于研究RNA結合蛋白和特定的RNA序列的相互作用,是轉錄因子研究的經典方法。金開瑞提供EMSA檢測技術服務,幫助您檢測DNA結合蛋白、RNA結合蛋白、特定的蛋白質,并可進行未知蛋白的鑒定。2)染色質免疫沉淀ChIP染色質免疫沉淀技術(Chromatin immunoprecipitation assay, ChIP)是將樣品中同抗體靶蛋白相互作用的DNA隨免疫復合物沉淀,是研究體內蛋白質與DNA相互作用的有力工具,利用該技術不僅可以檢測體內反式因子與DNA的動態作用,還可以用來研究組蛋白的各種共價修飾以及轉錄因子與基因表達的關系。3)RNA Pull Down / DNA Pull Down蛋白質與RNA的相互作用是許多細胞功能的核心,如蛋白質合成、mRNA組裝、病毒復制、細胞發育調控等。RNA Pull Down使用體外轉錄法標記生物素RNA探針,然后與胞漿蛋白提取液孵育,形成RNA-蛋白質復合物。該復合物可與鏈霉親和素標記的磁珠結合,從而與孵育液中的其他成分分離。復合物洗脫后,通過western blot實驗檢測特定的RNA結合蛋白是否與RNA相互作用。金開瑞現提供RNA pull down/ DNA Pull Down檢測技術服務, 使用特異性二抗進行檢測,能有效避免抗體重鏈對結果的信號干擾。并且金開瑞配套有世界上最先進的質譜儀,能顯著提升檢測效果。4)熒光素酶報告基因雙報告基因用于實驗系統中作相關的或成比例的檢測,通常一個報告基因作為內對照,使另一個報告基因的檢測均一化。理想的雙報告基因方法應該使用戶能夠以螢火蟲熒光素酶所具有的速度,靈敏和線性范圍對同一樣品中的兩個報告基因同時測定。金開瑞應用Promega 公司的雙熒光素酶報告基因檢測(DLR)系統,提供DLR檢測服務。5)酵母單雜交酵母單雜交技術最由酵母雙雜交技術發展而來的,通過對報告基因的表型檢測,分析DNA與蛋白之間的相互作用,以研究真核細胞內的基因表達調控。由于酵母單雜交方法檢測特定轉錄因子與順式作用元件專一性相互作用的敏感性和可靠性,現已被廣泛用于克隆細胞中含量微弱的、用生化手段難以純化的特定轉錄因子。RNA Pull Down 在博客最早就進行了系統地介紹,也在上一篇lncRNA-ZFAS1擴增促進肝癌轉移中應用到,實屬現階段研究蛋白質-核酸相互作用的常用手段。另外上一篇提到的RIP技術,雖然是利用抗原抗體特異性沉淀RNA-蛋白復合物,但最終目的是為了研究細胞內RNA 與蛋白結合情況,了解轉錄后調控網絡動態過程,助力發現miRNA 的調控靶點。如果您對上述服務方案感興趣,歡迎致電027-62431110垂詢相關實驗解決方案,或@qq3268908524索取最新的產品資料。[論壇]lncRNA研究之RNA pull-down問答集錦
2017-11-12 16:55:08
新開通了博客,說是寫點東西,剛好最近在學習理解非編碼RNA相關技術,簡單梳理了RNA pull down實驗中遇到的問題,可能不夠全面,也希望群友們給些建議,互相交流。lncRNA研究策略之RNA Pull-down實驗問答長鏈非編碼RNA(long non-coding RNA,lncRNA)是一類不編碼蛋白的RNA分子,長度在200bp以上;研究表明,lncRNA具有保守的二級結構,可以與蛋白、DNA和RNA相互作用,參與多種生物學過程的調控。隨著高通量測序技術的發展,越來越多的lncRNA被注釋,但是絕大多數的lncRNA的功能仍然不清楚,因此lncRNA的研究是一片非常廣闊的未知領域,具有極大的科研價值和意義。其中RNA Pull-down技術為lncRNA生物學功能的驗證起到了至關重要的作用。RNA Pull-down結合了體外轉錄與生物素標記、質譜技術,是研究非編碼RNA與蛋白質相互作用的一大利器。由于實驗程序較多、RNA的易降解性,加上一些不可預知的變量,因此實驗存在一些難度,常常導致實驗的失敗。對于試驗中常常出現的棘手的問題,http://http://www.genecreate.cn/RNA_pull_down/經過實踐,總結了一套行之有效的應對方策,現在一起來看看吧!
1、RNA Pull-down拉下蛋白PAGE電泳銀染條帶顏色淺且模糊?①選取的組織或細胞樣品其中互作蛋白表達量很低;②體外轉錄得到的RNA降解或者不是全長;③Pull-down實驗孵育時間不長;④其他2、PAGE電泳銀染背景深?①樣品雜蛋白太多;②銀染顯色時間過長;③其他
3、RNA Pull-down拉下蛋白銀染后看不到互作蛋白:①選取的RNA序列不對;②樣品中沒有表達互作的蛋白質;③互作蛋白濃度低,銀染圖肉眼觀察不到差異條帶;④其他更多建議:①lncRNA序列前期驗證要完善;②選擇相應蛋白質表達量較高的組織或細胞樣品;③體外轉錄選擇轉錄質量高的試劑盒,操作過程避免Rnase的污染;④保證RNA Pull-down實驗中探針標記以及探針與蛋白的結合充分進行(依據體外轉錄RNA的量,樣品裂解液蛋白濃度);⑤盡可能多將洗脫的蛋白電泳(同時可以避免顯色時間過長)其他原因,歡迎交流!如果您在RNA Pull-down實驗中遇到困惑,可以隨時@qq3268908524,下期我們再通過解讀一篇文獻中MS2 RIP、RNA Pull-down的技術應用,進一步探討,再見!探討lncRNA--ZFAS1的擴增促進肝癌轉移的機制
2017-11-12 16:09:31
參考文獻:Zhecheng Z, Li T, Xie J, et al. Amplification of long non-coding RNA ZFAS1 promotesmetastasis in hepatocellular carcinoma[J]. Cancer research, 2015: canres. 3721.2014.
基本點:MS2蛋白是MS2噬菌體的包膜蛋白,可以與噬菌體復制酶的5′端由19個堿基組成的莖環序列特異地結合。LncRNA--ZFAS1的擴增促進了肝癌細胞的轉移機制探討利用GEO數據庫及兩種lncRNA芯片數據,篩選得到肝癌組織中58個lncRNA個上調的和3個lncRNA下調,通過建立共表達網絡,確定了5個重要的lncRNA,其中上調最為明顯的是ZFAS1。因此后續的研究重點是ZFAS1在肝癌細胞中的表達模式、臨床意義、生物學功能。文獻中經典研究思路:(原諒我讀的文獻少,只是先解讀了主要的研究思路。)
研究者對ZFAS1促進肝癌細胞轉移的原因進行了探索。生物信息學分析表明,ZFAS1序列上有一個miR-150的結合位點,為了驗證這個結合位點,研究者構建了一個融合MS2莖環結構(即MS2結合蛋白互作位點MS2bs)的特殊ZFAS1重組轉錄本,該轉錄本能與MS2bp和GFP形成一個復合體,利用GFP抗體進行RIP實驗,對富集的miR-150進行qPCR驗證。結果表明,攜帶有WT型miR-150結合位點的ZFAS1重組轉錄本可以明顯富集miR-150,但mut型則無法富集miR-150。
 研究者們利用生物素標記的特異ZFAS1探針進行的RNA pull down也進一步證實了ZFAS1與miR-150的特異性結合。
研究者們利用生物素標記的特異ZFAS1探針進行的RNA pull down也進一步證實了ZFAS1與miR-150的特異性結合。 同時,也采用生物素標記的miR-150進行反向pull down,檢測miR-150是否能拉下ZFAS1,結果表明只有WT型miR-150可以富集ZFAS1,mut型則無法富集ZFAS1,綜合表明ZFAS1確實會作為一個ceRNA結合miR-150。
同時,也采用生物素標記的miR-150進行反向pull down,檢測miR-150是否能拉下ZFAS1,結果表明只有WT型miR-150可以富集ZFAS1,mut型則無法富集ZFAS1,綜合表明ZFAS1確實會作為一個ceRNA結合miR-150。 另外,研究者們也進一步證實,miR-150通過抑制內源ZEB1、MMP14、MMP16(腫瘤代謝中重要的調控因子)的表達減少細胞侵襲,ZFAS1通過影響miR-150對ZEB1、MMP14和MMP16三個基因的調控,造成三個基因表達上調,ZFAS1的沉默及過表達實驗也證明了ZFAS1通過競爭性結合miR-150,正調控ZEB1、MMP14、MMP16這三個基因的表達。(相關實驗設計和結果數據,能力、時間有限,后續再行補充更新該文章。)本項研究發現了肝癌細胞侵襲及轉移相關的一個重要lncRNA—ZFAS1,通過對ZFAS1在肝癌細胞中調控機制的進一步探索,結果表明ZFAS1通過影響miR-150調控腫瘤侵襲及轉移,ZFAS1的過表達促進了肝癌細胞的侵襲和轉移,為此,ZFAS1可能作為未來肝癌診斷及治療靶點,便于開發新的治療方法。(索取文獻&已解讀過的朋友也可@qq3268908524互相交流。我也是可以從網站上下載文獻的哦!文獻中采用的RIP、RNA pull down都屬于蛋白與核酸互作驗證常用技術方法,用于拉下RNA、蛋白質,而拉下蛋白的后續實驗目的通常聚焦到蛋白功能驗證上,常用手段之一--蛋白質組學,那么下期再會:iTRAQ經典技術分享)備注:本文章出于傳遞更多信息為目的解讀了該文獻部分內容,且已注明文獻、作者,不希望轉載的個人可與我聯系,會立即刪除處理。
另外,研究者們也進一步證實,miR-150通過抑制內源ZEB1、MMP14、MMP16(腫瘤代謝中重要的調控因子)的表達減少細胞侵襲,ZFAS1通過影響miR-150對ZEB1、MMP14和MMP16三個基因的調控,造成三個基因表達上調,ZFAS1的沉默及過表達實驗也證明了ZFAS1通過競爭性結合miR-150,正調控ZEB1、MMP14、MMP16這三個基因的表達。(相關實驗設計和結果數據,能力、時間有限,后續再行補充更新該文章。)本項研究發現了肝癌細胞侵襲及轉移相關的一個重要lncRNA—ZFAS1,通過對ZFAS1在肝癌細胞中調控機制的進一步探索,結果表明ZFAS1通過影響miR-150調控腫瘤侵襲及轉移,ZFAS1的過表達促進了肝癌細胞的侵襲和轉移,為此,ZFAS1可能作為未來肝癌診斷及治療靶點,便于開發新的治療方法。(索取文獻&已解讀過的朋友也可@qq3268908524互相交流。我也是可以從網站上下載文獻的哦!文獻中采用的RIP、RNA pull down都屬于蛋白與核酸互作驗證常用技術方法,用于拉下RNA、蛋白質,而拉下蛋白的后續實驗目的通常聚焦到蛋白功能驗證上,常用手段之一--蛋白質組學,那么下期再會:iTRAQ經典技術分享)備注:本文章出于傳遞更多信息為目的解讀了該文獻部分內容,且已注明文獻、作者,不希望轉載的個人可與我聯系,會立即刪除處理。

標題搜索
我的存檔
數據統計
- 訪問量: 0
- 日志數: 20
- 建立時間: 2017-11-09
- 更新時間: 2019-06-27
