采用Agilent 1200系列液相色譜系統進行藥物雜質分析
概述
引言
在日常質量控制和質量保證工作中,藥物中的雜質分析從初步篩選開始,以使用認證方法結束。在藥物價值傳遞鏈上,藥物雜質分析日益成為了一項具有挑戰性的任務。本匯編就高分離度快速液相色譜(RRLC)與質譜聯用如何用于藥物中雜質分析和定性提供一個指南,以提高您的工作效率。綜述了安捷倫最近的一些有關藥物中雜質分析的應用文摘。這些文摘以詳細摘要的形式介紹,若需深入閱讀全文,您可以從下面的網站下載:
www.agilent.com/chem/library。
藥物中的雜質
通常,從兩個方面關注藥物中的雜質:
? 化學方面包括對雜質的種類和鑒定,涉及到如何生成報告、如何設定適當的指標,以及描述分析步驟。
? 更重要的是病人的安全方面,當新藥上市時,這些病人將要使用最終產品。在這方面,比較研究和基因毒性測試變得越來越重要。
從法規機構如美國食品和藥品管理局(FDA)和EMEA等的觀點看,藥物中的雜質分成以下幾類:
? 有機雜質(與工藝和藥物相關)
? 無機雜質
? 殘留溶劑
本匯編提供的應用文摘系列“使用Agilent 1200系列液相色譜系統分析雜質”主要討論有機雜質:
? 第一部分:采用LC/MS鑒定雜質的結構(5989-5617EN)
? 第二部分:采用制備HPLC分離雜質(5989-5618EN)
? 第三部分:為方法開發快速確定分析條件(5989-5619EN)
? 第四部分:超快速方法的方法認證(5989-5620EN)
? 第五部分:附有完整步驟的QA/QC應用舉例(5989-5621EN)
在第15頁的附錄中有詳細的無機雜質和殘留溶劑文獻.
示例研究
本匯編中有5個應用文摘說明了在藥物開發和商品化過程中典型分析方法開發和QA/QC實驗室的分析流程,即,在藥物活性成分中分析測定與生產有關的雜質。
在研究的初期,采用高分離度快速液相色譜,通過加快分析速度,并與飛行時間質譜聯用,提供準確的質量信息,這就大大改進了分析過程。有機雜質來源有多種、濃度各不相同,它們最可能來源于藥物活性物質的合成和儲存過程,也可能來自最終藥物產品的制造工藝和/或儲存過程,可能來自于:
? 起始原材料
? 副產物
? 中間體
? 降解產物
? 試劑和配體,
也有來自包裝材料的。
根據FDA的指南“藥物中的雜質”,含量低于0.1%的雜質一般不必進行鑒定。然而,對于那些可能有異常作用、0.1 %以下濃度也可產生毒性或藥理作用的潛在雜質就應當進行鑒定。在所有情況下,雜質都應該定性。
雖然現實中一般把0.05 %到0.09 %的分析結果取近似值(即0.1 %),但本指南的目的,在確定是否對雜質進行鑒定時,這樣的值不應當近似為0.1 %。總之:為了減少潛在的副作用,需要對雜質進行靈敏的測定,而飛行時間(TOF)質譜能夠提供高靈敏度測定和準確質量鑒定,是鑒定痕量雜質的首選方法。
副產物分析
本出版物介紹的分析步驟主要涉及原料、副產物和中間體的分析,以及如何在鑒定過程中及早有效地檢測它們。關于包裝材料和有機揮發性雜質(OVI)分析的詳細說明可參看附錄。雜質分析遵循典型的方法開發流程,方法優化/轉移以及最后在GMP條件(圖1)下常規使用。

安捷倫科技長期以來致力于為制藥行業的分析化學家提供合適的儀器,以持續提高分析速度,并實現更高靈敏度的藥物分析。我們希望這個匯編是有用的,能夠提供保證病人安全的方法。
1. 采用LC/MS鑒定雜質的結構
引言
在現代制藥業、藥物開發和制造中,準確地鑒定微量雜質和副產物是極端重要的,因為雜質和副產物可能對人體有潛在的毒性。在藥物研發的各個階段分析雜質是非常重要的,而且是整個工藝的瓶頸。例如,本應用文摘討論了用高分離度快速液相色譜離子阱和電噴霧oaTOF質譜鑒定藥物合成過程中的副產物。
實驗
儀器
Agilent 1200系列高分離度快速液相系統(RRLC),
Agilent 6210飛行時間質譜
Agilent 6330離子阱質譜
方法
結果與討論

合成的主要產物是藥物和其非對映異構體,微量雜質A(圖1)。其它的可能雜質是降解產品(雜質B, C和D)以及合成的離析物(雜質E和F)。要在一個藥物化合物中發現所有可能的雜質,很重要的一點是使用幾種正交的分離技術,比如液相色譜、氣相色譜或薄層色譜。僅用LC/UV分析,合成中使用的離析物與它們的保留時間對比證明雜質F是3-bromanisole(圖1)。離析物雜質E用UV和ESI-MS都沒有檢測到。所以,又用GC-FLD分析了樣品,可以檢測到該化合物,并通過保留時間比較得到了證實(數據略)。為證實所有雜質的分子結構,進行了LC/MS-TOF分析,以測定準確的分子量并計算經驗分子式。該實驗可在低ppm下,以足夠準確的質量確證所有建議的分子式。要獲得有關雜質更詳細的結構信息,就要進行離子阱質譜分析(圖2)。非對映異構體雜質(A)在保留時間8.20 min檢測到,質量數為m/z 264.1。這一化合物的分子離子在MS/MS碎裂時由于失去水而產生碎片離子m/z246.1;在三級質譜(MS3)解離時,離子m/z 246.1由于失去甲氧基而產生主要離子m/z 215.1;失去甲基氨基產生離子m/z 202.1,以及與芐基陽離子相關的碎片離子m/z 121.1。


結論
我們論述了使用Agilent 1200系列高分離度快速液相色譜系統,1.8-μm的RRHT色譜柱,并與Agilent 6330離子阱和Agilent 6210 ESI TOF質譜聯用來對藥物的微量雜質進行檢測和結構鑒定。采用1200系列RRLC系統,在1.8-μm色譜柱上實現了檢測所有雜質需要的分離度。離子阱串聯MS/MS和MSn,可用于結構鑒定,ESI-TOF通過準確測定質量可用于初步分子式的確證。
2. 使用制備HPLC分離雜質
引言
雜質分析是一系列分析活動,目的是對藥物中的有機和無機雜質進行檢測、鑒定、結構解析和定量測定1。這一過程的第一個任務是檢測所有雜質,為此要使用各種正交分析技術,如氣相色譜,毛細管電泳和高壓液相色譜,包括聯用的MS技術,如離子阱和飛行時間(TOF)質譜。然而,即使采用最先進的質譜系統,解析所有化合物的結構也常常是不可能的。這些化合物必須分離和純化,然后用1H-和13C-NMR進行表征。這里介紹了一對用MS不能鑒定的化合物的分離和純化。
結果與討論
如Agilent應用文摘2中所述,采用離子阱和飛行時間質譜可以鑒定兩種雜質(A和D)。然而,另外兩種雜質(B和C)卻不能完全鑒定,所以采用制備HPLC進行分離以便實現進一步結構鑒定。分析方法2是針對雜質B和C的分離度以及較短的分離時間而優化的。方法放大之前,采用分析柱進行了上樣實驗。1.0444 mg原樣注射到ZORBAX SB-C18色譜柱上(4.6 x 150 mm, 5 μm)仍然可以實現雜質B和C的基線分離。
放大計算
在富集的樣品中,雜質B和C的濃度約為主要化合物的3 - 5 %,也就是說,在分析柱上每次進樣只可分離每種雜質約0.03 - 0.05 mg。因此,將分離放大到21.2 mm內徑的制備柱上,粗樣上樣量約22 mg,每次進樣可分離每種雜質約0.66 - 1.1 mg。這樣,不用十次進樣就能得到足以進行NMR分析的樣品量。
純化參數
使用化學工作站的Fraction Preview功能優化餾分收集參數。調用一個制備運行的色譜圖,調節餾分收集參數,如閾值(threshold)、上行斜率(up slope)、下行斜率(down slope)和上限閾值(upper threshold)等3,直到實現預期收集效果。在此情況下,時間窗口(9 - 14秒)內只基于閾值的收集就給出了最佳結果。圖1顯示了使用Fraction Preview優化參數得到的實際餾分收集運行結果。

將十次連續純化運行收集的雜質合并,雜質C(純度98.7 %)和雜質B(純度98.2 %)的量就足以用NMR進行結構分析(圖2)。

結論
從Agilent 1200系列RRLC系統上開發的高分離度分析方法開始,說明了兩種雜質的分離和純化。方法轉移到從Agilent 1200系列LC系統和從Agilent 1200系列制備系統是非常方便的,因為從Agilent的固定相既有用于分析柱的亞2微米的填料,也有用于制備柱的標準5-μm填料。所以,方法優化和上樣實驗之后的制備放大可以直接完成,而不必在制備柱上進一步優化方法。化學工作站的功能,如Fraction Preview和餾分自動收集很容易在短的時間內實現最佳純化。收集餾分的高純度和回收率使得雜質結構鑒定成了一件很容易的工作。
3. 快速篩選方法開發的條件
引言
“開發和認證新的分析方法成本高且費時”1所以,任何可減少單次分析運行時間的措施對整個分析過程都是有利的,但絕不能損失分離度、靈敏度、特別是可靠性。Agilent提供亞2微米填料粒徑度的色譜柱,以及1000倍以上使用壽命的高質量,這使得方法開發過程大為加快。開始時,常常要確定較寬的分離條件范圍,如固定相、流動相參數(% B, 緩沖液,pH,溶劑)和操作參數如梯度陡度、溫度等等,以確定最佳初始條件,然后進行微調。本應用文摘討論了使用Agilent 1200高分離度快速液相色譜(RRLC)系統和各種Agilent ZORBAX RRHT色譜柱來確定每次分析不超過4 min的分離條件,以及分離位置異構體化合物和非對映異構體的分離效率。
實驗
所有實驗均在Agilent 1200系列RRLC系統上完成,配置有SL型二元泵,溫控高效SL型自動進樣器,SL型柱恒溫箱和SL型二極管陣列檢測器。在條件篩選過程中,測試了16種不同的條件。下列Agilent ZORBAX色譜柱用作固定相:StableBond CN,StableBond C18, Extend C18, EclipseXDB C8和Eclipse XDB C18。流動相是HPLC級水和甲醇或乙腈,以及不同的添加劑。酸度條件為:0.2 %TFA(強溶劑為0.16 %),pH 1.92,弱酸條件是磷酸緩沖液,pH=6.02,最終的堿性條件是:0.2 %氨(pH=11.0)。色譜柱、流動相和pH的所有可能組合中,只試驗了合理的組合。初始篩選采用3.0 mm ID x 50 mm的色譜柱、低延遲體積的泵配置和5(L/6mm流通池的DAD。選擇一支4.6-mm ID的色譜柱進行方法微調,使用標準延遲體積的泵配置和13(L/10 mm的檢測器流通池。所有篩選試驗采用梯度為5 - 95 % B,溫度40℃。更詳細的情況請參閱第10頁的參考文獻2。
結果與討論

本方法開發的目的是提供一個快速而耐用的方法,用于藥物生產過程中雜質的定量,藥物為堿性鹽,pKa 9.4,易溶于水。從前面的實驗我們知道,采用其它技術如GC或TLC,而不是HPLC,就可以很容易地檢測到反應試劑。故將關注點放在非對映異構體A和區域去羥基化產物B和C、以及去甲基化產物D的分離上(見圖2的分子結構)。
在初期條件篩選過程中,使用富集了雜質的樣品,對于不同的固定相和流動相組合采用了5-95 % B的寬范圍梯度。正如所料,改變流動相和/或固定相,選擇性就發生變化,對色譜分離結果有顯著的影響。選擇了錯誤的條件可能導致錯誤的結論:樣品是很簡單的混合物。其它條件顯示有4個峰,最后的條件5個化合物都檢測到了。圖1給出了顯示選擇性變化作用的實例。在選擇微調的最佳初始條件進行微調時,只考慮分離度小于2的難分離物質對不超過1對的條件。最后,選擇了StableBond C18色譜柱進行進一步優化。微調首先將條件轉移到一支4.6mm ID的色譜柱上,這樣的柱內徑是制造業QA/QC環境優選的。然后,將梯度范圍變窄,還研究了溫度對微調方法的影響。最終方法對所有混合物可以得到大于3的分離度(圖2)。表1列出了分離含有低濃度(主成分的0.05 %)雜質的樣品的某些特征值。如參考文獻3和4所述,開發的方法能滿足標準認證程序以及在制藥QA/QC條件下應用的所有要求。
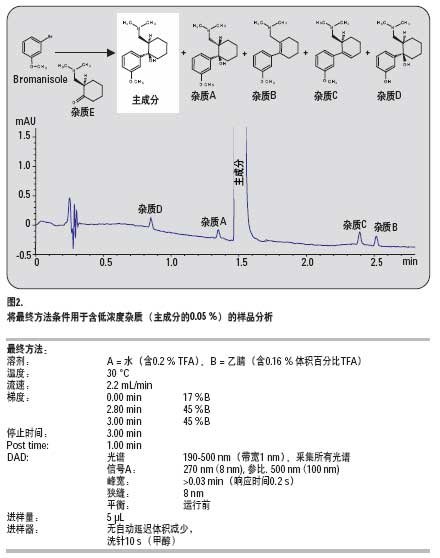

結論
使用Agilent 1200系列RRLC系統和亞2微米粒徑填料的色譜柱進行條件篩選和微調可以減少活性藥物組分中難分離的非對映異構體和位置異構體雜質的方法開發時間。由于需要很短的篩選運行周期(4.5 min),一天之內可以試驗很多組條件(包括重復和空白運行)。其它的微調再用半天時間,所以,一天半之后就可提供一個方法用于后續的方法認證,由于采用最終方法每次單個運行的時間只有4 min,因此也會明顯減少方法認證的時間。
4. 快速LC方法的方法認證
引言
為了保證分析結果符合法規要求,在滿足法規依從性的分析實驗室必須對他們的方法進行認證。此外,盡管他們使用不同的儀器。不同實驗室的不同用戶所得結果應是可比的,所以,方法必須盡可能穩定可靠,以對誤差進行補償,因為不同的用戶在相同或不同的儀器上運行相同的分析時可能產生誤差。這里介紹的認證程序是基于美國藥典(USP),ICH指南Q2B1,2以及FDA指南的要求4,5,這些要求為世界各國所認可,并被制藥、食品、環境和化工分析實驗室所采用。下面介紹分析一個主成分及其4個雜質的快速LC方法的認證,圖1給出了典型色譜圖。

認證程序
在預認證實驗之后,設置了表1所列的認證協議。測試并給出了專屬性3。沒有采取進一步的樣品制備步驟。稱取樣品化合物并溶于水。設定了組成的限量并與實測值比較以確定通過或失敗(表2)。雜質的分析結果列于表3。



結論
為分析主成分和4種雜質開發了快速LC方法。該方法的認證是成功的,滿足了涉及精度、線性、準確度和穩定可靠性的所有要求。這說明快速LC方法可以用于QA/QC實驗室,且符合USP/ICH推薦的要求。快速LC方法提供了同樣的數據質量,而且又有較高的樣品通量。
5. QA/QC應用舉例,采用快速LC方法實現高通量樣品分析
引言
已經開發了一個快速分析LC方法,在對這一快速分析方法認證之后,下一步就是將這一方法轉移到QA/QC實驗室。通常,詳細的標準操作規程(SOP)保證分析過程中出錯和誤解的幾率盡可能小。下面是一個標準操作規程的例子,包括系統適用性測試、序列和結果通過認證的指標。Agilent 1200系列高分離度快速液相色譜(RRLC)系統用來進行序列分析、系統適用性檢測報告、樣品和校準混合物的分析。色譜條件是基于以前的應用文摘所開發認證的LC方法,參見文獻1和2。約5 min的樣品分析周期應當大大提高了樣品通量,進而降低了分析成本。下面是簡化的SOP舉例,并給出了結果舉例。
性能測試
要確定一個樣品是通過還是失敗需要多次測試。必須使用校準混合物和對照樣品,以及含有用來稀釋樣品的溶劑的空白樣品。表1概要列出了測定樣品分析的精度、靈敏度、分離度和其他性能參數的要求。基于該表定義的要求,在安捷倫化學工作站軟件中設置了一個分析序列表(表2)。這個序列的開始是適用性測試樣品,在3個樣品和一個對照樣品分析之前和之后是校準運行,在這些運行之間要注射純水樣品以保證沒有記憶效應或鬼峰,因為這些會干擾下一次分析。


測試結果舉例
作為一個例子,我們介紹系統適用性測試規程。要測試系統是否滿足方法要求,制備了主成分和雜質A、B、C、D的溶液,濃度為4 - 10 μg/mL。每天第一次進樣之前都要分析這個溶液。
測試的參數和極限值設置:
? 峰面積的精度必須< 2 % rsd
? 保留時間的精度必須< 0.5 % rsd
? 所有峰的分離度必須> 2
? 最大的半峰寬必須< 0.08 min
? k′必須5 < k′< 25
? 所有峰的信噪比必須> 50
系統適用性樣品的測試結果列于表2。滿足了所有的極限指標。以同樣的方法,評價了校準標樣和對照樣品的參數指標。所有測試都滿足了極限指標,被分析樣品也通過了極限指標,3個樣品的雜質含量均沒有超過0.5 %的允許極限值。
結論
分析QA/QC部門面臨著分析樣品數量的逐漸增多以及對數據質量和可靠性越來越高的要求。一般來說,為保證定性和定量數據的正確性,一個樣品需要運行10-15次。本文完整地認證的快速LC方法有助于顯著提高樣品分析通量,而不損失數據的質量。所用序列包含47次分析運行,包括30 min的系統適用性測試,共用3.9小時。若使用一個樣品分析周期為20 min左右的方法,大致需要15.7小時。這說明快速LC方法明顯提高了樣品通量。
溶劑的節省也非常明顯。采用20 min的樣品分析周期,47次運行,流速為2.2 mL/min,需要溶劑2068 mL。而采用本文的快速LC方法,5 min的樣品分析周期,47次運行只需要517 mL溶劑。
每次分析的成本因而也大大下降為大約原來的1/5~1/4。見表3的舉例。

方法的重新認證可能需要2周時間,花費4000美元。在我們的例子中,在約500次分析運行之后,將方法升級為快速方法在成本上經濟實惠。
附錄
雜質分析
“Impurity profiling with the Agilent 1200 Series LC system ?Part 1: Structure Elucidation of Impurities with LC/MS”, Agilent Application Note, publication number 5989-5617EN, 2006.
“Impurity profiling with the Agilent 1200 Series LC System ?Part 2: Isolation of Impurities with Preparative HPLC”, Agilent Application Note, publication number 5989-5618EN, 2006.
“Impurity profiling with the Agilent 1200 Series LC system ?Part 3: Rapid Condition Scouting for Method Development”, Agilent Application Note, publication number 5989-5619EN, 2006.
“Impurity profiling with theAgilent 1200 Series LC System,Part 4: Method Validation of Ultrafast Method” Agilent Application Note, publication number 5989-5620EN, 2006.
“Impurity Profiling with the Agilent 1200 Series LC System Part 5: V.QA/QC Application Example with Complete Sequencing” Agilent Application Note, publication number 5989-5621EN, 2006.
行業指南
“Guidance for Industry NDAs: Impurities in Drug Substances,” U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research(CDER) CMC, February 2000.
“Guidance for Industry Q3B(R2) Impurities in New Drug Products,” U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) ICH Revision 2, July 2006.
殘留溶劑
“The Determination of Residual Solvents in Pharmaceuticals Using the Agilent G1888 Network Headspace Sampler”, Agilent Application Note, publication number 5989-1263EN, 2004.
“The Determination of Residual Solvents in Pharmaceuticals Using the Agilent G1888 Headspace/6890N GC/5975 Inert MSD System”, Agilent Application Note, publication number 5989-3196EN, 2005.
“The Determination of Extractables and Leachables in Pharmaceutical Packaging Materials Using Headspace GC/MS”,Agilent Application Note, publication number 5989-5494EN,2006.
無機雜質
“Evaluation of conventional ICPMS and ORS-ICP-MS for Analysis of Traditional Chinese Medicines”,Agilent Application Note, publication number 5989-2570EN,2005.
“Determination of Toxic Elements in Traditional Chinese Medicine Using Inductively Coupled Plasma Mass Spectrometry”, Agilent Application Note, publication number, 2006.
“Routine Pharmaceutical Analysis:The Indispensable Solutions Guide to Proven Applications and Best Practices” CD, Agilent publication number 5989-5366EN, 2006.

相關閱讀:
- Agilent 7890A/NPD/5975C/DRS GC/MSD在法醫毒物快速分析中的應用 (UUBird, 2008-6-16)
- 平行氣相色譜系統實現完整的煉廠氣分析 (UUBird, 2008-6-16)
- 用Agilent 1120一體式液相色譜儀同時測定撲熱息痛_雙氯芬酸和布洛芬的方法建立和驗證 (UUBird, 2008-6-16)
- 用高分離度快速液相色譜-UV和MS檢測建立可靠的中藥制劑質控方法 (UUBird, 2008-6-16)
- 用LC/TOF-MS鑒定環氧酚類食品罐頭涂層中未知的反應副產物和污染物 (UUBird, 2008-6-16)
- 同位素稀釋氣相色譜/質譜測定環境樣品中的丁基錫化合物 (UUBird, 2008-6-16)
- 用配置亞2 微米粒徑色譜柱的液相色譜儀改進對生物樣品中代謝物的分離 (UUBird, 2008-6-16)

