







仿制藥中有關物質研究(一)
? 本文在總結多年審評仿制藥研發資料經驗、長期從事藥品檢驗的感受及藥品品質評價法與臨床療效間的相關認知等基礎上,詳盡闡述了進行仿制藥研發時有關物質的研究思路,宏觀地解讀了藥物臨床使用時雜質的副作用。
現今,有關物質研究已成為仿制藥研發的重中之重。自2007年我國修訂《藥品注冊管理辦法》以來,國家藥品審評中心發布了大量有關物質研究指導原則,各國藥典、進口質量標準也對有關物質制訂了翔實充分的質控指標。近年來,國內注射劑不良反應事件層出不窮,眾人也將雜質歸結為主要因素之一。基于以上背景,雜質研究成為了業內關注的焦點。
制訂有關物質檢查項的原則
原料藥質量標準
原料藥質量標準通常需制訂。即便該原料藥穩定性良好,在效期內雜質無任何增加/變化,質量標準中也應制訂有關物質檢查項。此舉是通過對合成工藝雜質的控制與評估,來保證批次間原料藥生產穩定性與質量均一性。
制劑質量標準
應研究“原料藥制成0天制劑”和“0天制劑在效期內流通”兩個環節的雜質變化情況。若一個環節有變化,就需擬定有關物質檢查項;而均無變化可不擬定。
雜質譜研究邏輯樹
目前,全球藥品研發皆提出了“雜質譜研究”理念,仿制制劑與原研制劑劑型不同時可參照該思路。由于臨床使用的是制劑,仿制制劑的降解雜質通常與原研制劑相同,而仿制原料藥的工藝雜質與原研原料藥可能不一致,因此無需對各國藥典原料藥質量標準項下羅列的所有雜質逐一研究,僅關注其中與制劑相同的降解雜質即可。
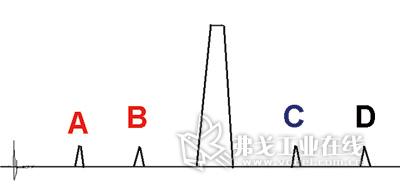
圖1. 原研制劑雜質譜測定結果。
購買數批原研制劑
無論是仿制藥研發還是品質評價,皆應獲取至少3個不同時間段批號的原研制劑,并最好有近效期的樣品,以知曉該時間點時雜質的降解情況和含量。隨后,取最新批號樣品進行加速或長期試驗,進一步觀測雜質降解情況和增長速率,同市場流通樣品做綜合比較分析。仿制藥研發工作建議在原研品上市后啟動,一年購買1個批號,測定其多條溶出曲線和雜質譜,并觀測近效期時這些指標的變化情況,以更科學合理地指導屆時自我仿制品的開發工作。
六類/五類仿制藥研發思路
解讀既有質量標準
針對此類仿制藥研發,建議查詢所有可及的制劑與原料藥質量標準;著重關注制劑質量標準,解讀試驗條件、雜質種類、雜質質控限度等,原料藥的質量標準應著重關注與制劑相同的主成分降解雜質。
研發時僅是借用既有質量標準的檢測色譜條件和目標降解雜質限度值,至于雜質種類和個數等是根據自我仿制品的研發實際情況針對性地研究和擬定。不同生產單位可能會采用不同的原料藥制備工藝、制劑的處方工藝,從而導致產品質量控制方法的不同。需遵循仿品種而非仿標準的原則,以研制產品與已上市產品安全性、有效性一致為目標,針對具體品種制定個性化注冊標準。
實踐既有色譜條件并予以優化
建議采用25cm普通色譜柱,以更有效增強分離效能。不推薦使用超高速液相,因該試驗所受干擾因素較多,難重現,不便推廣。對于反相色譜檢測,可適當減少有機相比例,使主成分保留時間延長、各峰分離度增加,或至檢出的雜質數不再增加。供試品溶液的主成分保留時間推薦在15~30min間,全部檢測時間約在45~90min。既可將各種雜質盡可能分離,又可通過調整峰寬與斜率,使保留時間較長的雜質峰即便展寬也可積分出,不會導致漏檢。
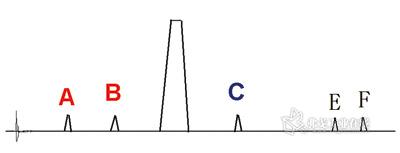
圖2. 仿制原料藥和仿制制劑雜質譜測定結果。
流速與柱溫以滿足上述保留時間為宜,流速無需拘泥于0.8~1.2ml/min間;柱溫略高于室溫,有利于延緩色譜柱使用壽命。儀器配置推薦具有自動進樣裝置的色譜儀。梯度洗脫時流動相配制最科學方式為A相為高比例水相-低比例有機相,B相為低比例水相-高比例有機相;其次是A相為高比例水相-低比例有機相,B相為純有機相。
梯度洗脫程序皆可靈活地更改,如延長洗脫時間、調整最終兩相比例、延緩每分鐘變化率等,已達到分離自我樣品的目的。采用最終優化的色譜條件,能夠將仿制原料藥/制劑的各工藝雜質/降解雜質/輔料干擾等與主成分良好分離。此時的系統適用性試驗用溶液應將雜質:主成分配制成1:100濃度來驗證專屬性。同時,還應進行強制降解試驗,用以驗證各種強破壞試驗條件下,主成分與產生雜質的分離檢測結果,應特別關注DAD檢測條件下主峰純度,如不純,則應適當調整色譜條件參數,將雜質分離出,無需檢測雜質峰純度。
測定原研制劑雜質譜
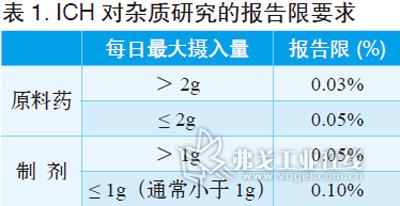
在對供試品溶液積分時,其中一項重要參數就是最小峰面積的設定,即雜質報告限,ICH在原料藥與制劑中雜質研究指導原則中規定如表1所示。
除非既有質量標準中有低于0.05%限度的雜質要求,通常可根據以上規定予以設定。如僅進行仿制制劑研發,建議積分限度設定為0.10%;如要同時進行仿制原料藥研發,遵循雜質譜研究邏輯樹,建議將積分限度設定為0.05%。這也同時考慮了方法學中最低檢測限和最低定量限等指標的驗證。
雜質測定結果的記錄保留到小數點后兩位即可,在上述條件下測定原研制劑雜質譜。如未能獲得臨近效期的樣品,可采用最新批號樣品進行影響因素試驗,還應進行加速試驗以觀測雜質變化情況,尤關注不斷增加的雜質。往往在加速試驗3個月時,便可初現端倪。測定結果如圖1所示。
雜質A和B隨時間延長、含量不斷增加;加速試驗也證明,結果分別為0.15%→0.50%和0.20%→0.68%,故確定為主成分降解雜質。經核對原研制劑質量標準,通過相對保留時間定位推算確定為某代號雜質;再根據既有質量標準,知曉兩雜質限度分別為0.7%和1.0%(數字皆為舉例、以下同)。雜質C和D隨時間延長、含量不變化;加速試驗也證明不增加,故推斷為主成分原料藥工藝雜質或輔料峰。檢測結果分別在0.33%~0.35%和0.07%~0.08%間波動。
測定仿制原料藥雜質譜
美國FDA發布的《仿制制劑雜質研究指導原則》中指明:ICH在新原料藥與新制劑中雜質研究指導原則中的許多建議完全適用于仿制藥研究,故積分限度參照“最小峰面積的設定”項下依據設定為0.05%。測定結果如圖2所示。
對于雜質A和B,研發要求3批樣品0天時的含量不應高于原研制劑起始點含量(即0.15和0.20%),加速試驗證實不斷增加,但增加速度不高于原研制劑,且未超出限度值。雜質C經研究確證為合成中間體、即合成工藝中引入的雜質。研發要求3批樣品0天時的含量不大于原研制劑,加速試驗證實不增加。雜質D未檢出,說明該雜質為原研制劑特有雜質,勿需研究。對于仿制原料藥中特有雜質E和F,3批樣品0天時含量均分別為0.12%和0.07%。經驗證,雜質E為合成中間體雜質,因合成路徑與原研原料藥不同,加速試驗證實不增加;雜質F增加、為新增降解雜質,但至加速試驗6個月時未超出0.2%。
根據表2,由于以上兩雜質均不過0.1%就無需鑒定;但考慮到最終臨床使用的是制劑,故可再根據制劑要求,放寬至0.2%(或0.5%,以下同)。即仿制原料藥中的特有雜質只要最終含量未超過0.2%(主成分日劑量10mg~2g),即可不進行結構確認等的進一步研究。
當特有雜質量超出0.2%,建議對原料藥合成工藝進行完善與優化,減少含量至鑒定閾值以下,否則就需進行結構確認→定量檢測→甚至動物毒理試驗推算該雜質限度值的深入研究。
如研發制劑涉及的原料藥為外購時,制劑生產企業可要求原料藥企業按既定色譜條件檢測,若達不到要求,應進行精制優化,直至符合規定。
測定仿制制劑雜質譜
測定結果如圖2所示。對于雜質A和B,研發要求三批樣品0天時的含量不應高于原研制劑起始點含量,加速試驗證實不斷增加,但增加速率不快于原研制劑,且最終時間點含量未超出既有質量標準的限度值。
對于雜質C,要求結果同仿制原料藥,加速試驗證實不增加。由于原研制劑在該雜質含量的前提下臨床已應用多年,安全可靠,且含量未超過原研制劑,故仿制制劑與原料藥就無需再深入研究該雜質。推薦采用液質聯用色譜儀做一定性試驗予以驗證,或用兩種以上色譜系統驗證為同一物質即可。雜質D未檢出,同仿制原料藥。雜質E和F的研發要求為加速試驗證實雜質E不增加。雜質F增加、但至加速試驗6個月時未超出0.2%。由于均未過鑒定限,故無需進行結構確認等進一步研究。
-
焦點事件

-
產品技術

-
產品技術

-
產品技術

-
焦點事件
-
焦點事件









