2020年版中國藥典-四部:9102 藥品雜質分析指導原則
9102 藥品雜質分析指導原則
本原則用于指導化學合成的原料藥及其制劑的雜質分析,并供藥品研究、生產、質量標準起草和修訂參考。本原則不涵蓋生物/生物技術制品、肽、寡聚核苷酸、放射性藥品、發酵產品與其半合成產品、中藥和來源于動植物的粗制品。
雜質是藥品的關鍵質量屬性,可影響產品的安全性和有效性。藥品質量標準中的雜質系指在按照經國家藥品監督管理部門依法審查批準的工藝和原輔料生產的藥品中,由其生產工藝或原料帶入的雜質,或在貯存過程中產生的雜質,不包括變更生產工藝或變更原輔料而產生的新雜質,也不包括摻入或污染的外來物質。若藥品生產企業變更生產工藝或原輔料引入新的雜質,則需要對原質量標準進行修訂,并依法向藥品監督管理部門申報批準。藥品中不得摻入其組分以外的物質或污染藥品。對于假藥和劣藥,必要時應根據具體情況,采用合適的且經過驗證的分析方法予以檢測。
1. 雜質的分類
藥品雜質通常分為:有機雜質、無機雜質、殘留溶劑。有機雜質可在藥品的生產或貯存中引入,也可由藥物與輔料或包裝結構的相互作用產生,這些雜質可能是已鑒定或者未鑒定的、揮發性的或非揮發性的,包括起始物、副產物、中間體、降解產物、試劑、配位體和催化劑;其中化學結構與活性成分類似或具淵源關系的有機雜質,通常稱為有關物質。無機雜質可能來源于生產過程,如反應試劑、配位體、催化劑、元素雜質、無機鹽和其他物質(例如:過濾介質,活性炭等),一般是已知和確定的。藥品中的殘留溶劑系指原料藥或輔料的生產中,以及制劑制備過程中使用的,但在工藝操作過程中未能完全去除的有機溶劑,一般具有已知的毒性。
由于雜質的種類較多,所以,藥品質量標準中檢查項下雜質的項目名稱,應根據國家藥典委員會編寫的《國家藥品標準工作手冊》的要求進行規范。如有機雜質的項目名稱可參考下列原則選用。
(1)檢查對象明確為某一物質時,以該雜質的化學名作為檢查項目名稱,如磷酸可待因中的“嗎啡",氯貝丁酯中的“對氯酚”,鹽酸苯海索中的“哌啶苯丙酮”,鹽酸林可霉素中的“林可霉素B”和胰蛋白酶中的“糜蛋白酶"等。如果該雜質的化學名太長,又無通用的簡稱,可參考螺內酯項下的“巰基化合物”、腎上腺素中的“酮體”、鹽酸地芬尼多中的“烯化合物”等,選用相宜的名稱。在質量標準起草說明中應寫明已明確雜質的結構式。
(2)檢查對象不能明確為某一單一物質,而又僅知為某一類物質時,則其檢查項目名稱可采用“其他甾體”“其他生物堿”“其他氨基酸”“還原糖”“脂肪酸”“芳香第一胺”等。
(3)未知雜質,可根據雜質性質選用檢查項目名稱,如“雜質吸光度”“易氧化物”“易炭化物”“不揮發物”“揮發性雜質”等。
2. 質量標準中雜質檢查項目的確定
新原料藥和新制劑中的雜質,應按我國新藥申報有關要求和ICH新原料藥中的雜質(Q3A)和新制劑中的雜質(Q3B)指導原則進行研究,必要時對雜質和降解產物進行安全性評價。新藥研制部門對在合成、純化和貯存中實際存在的雜質和潛在的雜質,應釆用有效的分離分析方法進行檢測。對于表觀含量在表1鑒定閾值及以上的單個雜質和在鑒定閾值以下但具強烈生物作用的單個雜質或毒性雜質,予以定性或確證其結構。對在藥品穩定性試驗中出現的降解產物,也應按上述要求進行研究。新藥質量標準中的雜質檢查項目應包括經質量研究和穩定性考察檢出的以及在批量生產中出現的雜質和降解產物,并需制定相應的檢查限度。除降解產物和毒性雜質外,原料藥中已控制的雜質,制劑中一般不再控制。原料藥和制劑中的無機雜質,應根據其生產工藝、起始原料情況確定檢查項目,但對于毒性無機雜質,應在質量標準中規定其檢查項。藥品雜質的報告、鑒定和確證閾值參照ICH新原料藥中的雜質(Q3A)和新制劑中的雜質(Q3B)指導原則(表1)。若制定的閾值高于表1閾值,則需進行科學評估;若雜質的毒性很大,應制定更低閾值。
在仿制藥的研制和生產中,如發現其雜質譜與其原研藥不同或與已有法定質量標準規定不同,需增加新的雜質檢查項目時,也應按上述方法進行研究,申報新的質量標準或對原質量標準進行修訂,并報藥品監督管理部門審批。
多組分藥物中共存的異構體一般不作為雜質檢查項目,必要時,在質量標準中規定其比例,以保證生產用與申報注冊時的原料藥一致性。但當共存物質具有毒性時,應作為毒性雜質進行檢查。而在單一對映異構體藥品中,可能共存的其他對映異構體和非對映異構體應作為雜質檢查。
藥品多晶型雜質,應參照本藥典藥品晶型研究及晶型質量控制指導原則(指導原則9015),確定檢查項目。
具有遺傳毒性的雜質(又稱基因毒性雜質),應參照ICH評估和控制藥品中DNA反應性(致突變)雜質以降低潛在致癌風險指導原則(M7)進行研究,并確定檢查項目。
無機雜質參照ICH元素雜質指導原則(Q3D)進行研究,并確定檢查項目。

a 取限度低者
報告閾值(reporting threshold):超出此閾值的雜質均應在檢測報告中報告具體的檢測數據。鑒定閾值(identifica tionthreshold):超出此閾值的雜質均應進行定性分析,確定其化學結構。確證閾值(qualifica tionthreshold):超出此閾值的雜質均應基于其生物安全性評估數據,確定控制限度。TDI:藥品雜質的每日總攝入量(total daily intake)。
殘留溶劑,應根據生產工藝中所用有機溶劑及其殘留情況,參照本藥典殘留溶劑測定法(通則0861)和ICH殘留溶劑指導原則(Q3C),確定檢查項目。
3. 雜質檢查分析方法
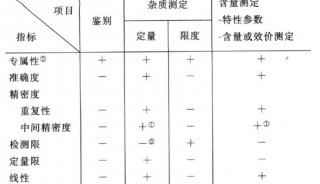
雜質檢查應盡量采用現代分離分析手段,用于雜質檢測和定量測定的分析方法須按照本藥典分析方法驗證指導原則(指導原則9101)和ICH指導原則(Q2)進行驗證。尤為重要的是,應能證明分析方法具有檢測雜質的專屬性。
研究時,應采用幾種不同的分離分析方法或不同檢測條件以便比對結果,選擇較佳的方法作為列入質量標準的檢查方法。雜質檢查分析方法的建立,應考慮普遍適用性,所用的儀器和實驗材料應容易獲得。對于特殊實驗材料,應在質量標準中寫明。在雜質分析的研究階段,將可能存在的雜質、強制降解產物,分別或加入主成分中,配制供試溶液進行色譜分析,優化色譜條件,確定適用性要求,保證方法專屬、靈敏。
雜質研究中,應進行雜質的分離純化制備或合成制備,以供進行安全性和質量研究用。對確實無法獲得的雜質,研制部門在藥品質量研究資料和藥品質量標準起草說明中應寫明理由。
在采用現代色譜技術對雜質進行分離分析的情況下,對特定雜質中的已知雜質和毒性雜質,應使用雜質對照品進行定位;如無法獲得雜質對照品時,可用相對保留值進行定位。雜質含量可按照色譜法等測定。
對于對映異構體雜質的檢測多采用手性色譜法或其他立體選擇性方法,應用最為廣泛的是手性高效液相色譜法。對于對映異構體雜質檢查方法的驗證,立體選擇性是實驗考察的重點。當對映異構體雜質的出峰順序在前,母體藥品在后,則有利于兩者的分離和提高檢測靈敏度。由于手性色譜法不能直接反映手性藥品的光學活性,需要與旋光度或比旋度測定相互補充,以有效控制手性藥品的質量。對消旋體藥物的質量標準,必要時亦可以設旋光度檢查項目。
由于采用色譜法進行雜質限度檢查時,受色譜參數設置值的影響較大,有關操作注意事項應在起草說明中寫明,必要時,可在質量標準中予以規定。
4. 雜質的限度
藥品質量標準對毒性雜質和毒性殘留有機溶劑應嚴格規定限度。雜質限度的制訂可參考本藥典和ICH相關指導原則的要求,考慮如下因素:雜質及含一定限量雜質藥品的毒理學和藥效學研究數據,原料藥的來源,給藥途徑,每日劑量,給藥人群,治療周期等。
原料藥和制劑質量標準應包括如下。
(1)每種特定的已鑒定雜質。
(2)每種特定的未鑒定雜質。
(3)任何不超過鑒定閾值的非特定雜質。
(4)雜質總量(所有超過報告閾值的特定和非特定雜質或降解產物的總和)。
藥品雜質鑒定與質控的決策樹如下。