引言
提交到華盛頓州毒物學實驗室的血樣中,20% 以上都對阿片類化合物、可卡因代謝物的免疫篩選呈陽性。死亡調查和藥物濫用所致拘留占大多數,也包含少數性侵犯。各案例之間的藥物濃度有很大差別,待測物呈現出多種不同的組合。理想的確認分析應該可以在一次血樣分析中測定所有阿片類化合物、可卡因和可卡因代謝物,并具有高靈敏度和寬線性動態范圍。直到最近,氣相色譜-質譜還是這種確認的行業標準,但是這種方法要對樣品進行衍生化,甚至需要兩次衍生[1]。
在華盛頓州毒物學實驗室,我們用安捷倫MSD SL 和新的ZORBAX Eclipse Plus C18 柱,采用液相色譜/質譜法(LC/MS),對幾千個案例中的阿片類化合物、可卡因和可卡因代謝物進行了分析。與我們以前用的GC/MS 方法相比,這一方法有許多優點,包括樣品處理更簡便、靈敏度更高,可以在一次分析中檢測更寬范圍的阿片類化合物。
實驗部分
方法
提取
凈化篩分萃取柱(聯合化學技術公司,CSDAU206)預處理
1 x 3 mL 甲醇
1 x 3 mL 去離子水
1 x 3 mL 0.1 M KH2PO4
血樣制備
(標準品:加入工作標準液并先干燥)
50 μL 內標(乙基嗎啡2 μg/mL)
1 mL 血液
3 mL 0.1 M KH2PO4
旋渦混合并離心,2,500 rpm 15 分鐘 以1 到2 mL/min 的流速將稀釋、離心后的血液加到平衡好的凈化柱上。
洗柱
1 x 3 mL 蒸餾水
1 x 3 mL 0.1 N HCl
1 x 3 mL 甲醇
在高真空下干燥10 分鐘
洗脫
1 x 3 mL CH2Cl2/異丙醇/NH4OH (72/26/2)(每天新鮮配制/現用現配)
蒸發@ 50o(~ 20 分鐘)并用100 μL 1%冰醋酸復溶
色譜和儀器條件

MS條件

結果和討論
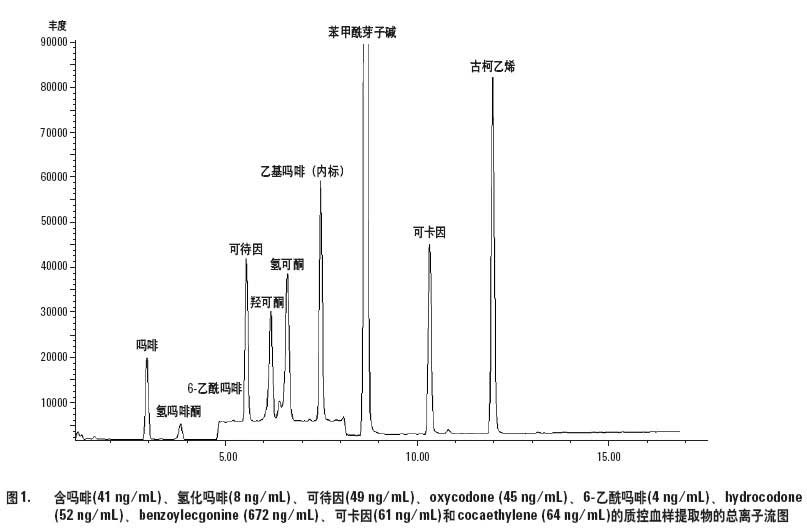
表2 列出本方法中化合物的保留時間和所用的離子。安捷倫應用報告988-4805EN[2]中給出了這些化合物的化學結構。將原始數據文件從LC/MSD計算機轉入另一個計算機,用安捷倫MSD化學工作站進行數據分析(Agilent LC/MS 數據文件與MSD化學工作站完全兼容)。對每個待測物,有一個目標質量數是加質子形成的準分子離子(M+H)。為了使碰撞誘導裂解能得到足夠的確認離子豐度,采用了較高的裂解器電壓。每個化合物至少監測兩個質量數,每個離子比值的限定范圍設定在± 25% [3]。當一個化合物監測兩個離子時,將同位素離子(M+2)作為第三個離子,但其不能和代表目標分子已知碎片的確認離子一樣,提供足夠多的信息。在這種情況下,形成的加鈉離子(M+H+22),就不一定能作為確認離子。對照血樣提取物的色譜圖見圖1。
表3列出了本方法的檢出限、定量限和線性,以及6個月內采集的質控數據。實驗室的方針是將血液藥物控制的合格范圍設置在內部測定平均值的± 20%。所有常規測試分析物的校正曲線相關系數(r2) ≥ 0.990。除嗎啡-3-葡萄糖酸和嗎啡-6-葡萄糖酸外,所有分析物的回收率為均大于90%。嗎啡葡萄糖酸的回收率較低(~1%)。提取物進樣后沒有洗針時,前一針進樣的交叉污染比較明顯,但方法中加入洗針后,濃度高達10,000 ng/mL 或更大,都不再有交叉污染。


我們的方法是在Pichini 等人[4]所報道方法的基礎上建立的。我們嘗試加入阿片類化合物,不改動其分析步驟,但羥可酮、氫嗎啡酮和氫可酮出現嚴重的不對稱峰。這一問題在以前的文獻中已有報道,認為是與流動相成分形成多種加合產物/復合物的結果[5]。使用高效Eclipse Plus 柱,采用較高的溫度(60 ℃),有效地改善了這些有問題分析物的峰形(圖1)。
用一份提取物分析盡可能多的阿片類化合物,不僅大大節省了時間和經費,而且還有其它好處。用這一方法可進行細胞色素P450 2D6 代謝產生的可待因、氫可酮、羥可酮的次級活性代謝產物(分別來自嗎啡、氫嗎啡酮和羥嗎啡酮[6])的常規監測。這些活性代謝物的信息可能對評價總的阿片/麻醉作用有幫助,也有助于區分急性和慢性藥物接觸。這種LC/MS 方法,可以在下列三種不同情況下檢測到氫嗎啡酮:(1)服用氫嗎啡酮后;(2)作為氫可酮的次級代謝物;(3)大劑量服用嗎啡后的次級代謝物[7]。
本方法中,選擇乙基嗎啡作為內標,因為我們用單級四極桿儀器測定的某些氘代內標,碎裂時產生和其同系列目標化合物一樣的離子。如果本方法使用的是串聯LC/MS 系統,很多氘代內標都可以用,得到的結果甚至可能比這里報告的準確度和精密度更高。
羥可酮和嗎啡-3-葡萄糖酸苷、嗎啡-6-葡萄糖酸苷的分析還只用于研究。盡管回收率較低,但和嗎啡一起測定嗎啡-3 和嗎啡-6-葡萄糖酸苷對判斷急慢性藥物攝取差異頗有價值。在一起與嗎啡相關的致死案例中,一位尋求刺激的少年服用了一位年長婦女開具的持續釋放嗎啡的未知制劑。LC/MS分析顯示尸檢血液中嗎啡濃度超過700 ng/mL,但嗎啡葡萄糖酸苷的濃度卻較低。相反,對長期攝入嗎啡的癌癥死亡病人尸檢,嗎啡葡萄糖酸苷的濃度通常要比前體藥物濃度高一個數量級。增加本方法洗脫溶劑中異丙醇的比例,可以提高嗎啡葡萄糖酸苷的回收率。用更簡單的疏水固相萃取柱提取[2],而不是用本方法報道的疏水/陽離子交換柱,嗎啡葡萄糖酸苷的回收率很高,但缺點是背景信號增高,柱壽命降低。另一種能夠滿足要求的提取方法是采用聚合固相柱,用5% 氫氧化銨甲醇洗脫,嗎啡及其葡萄糖酸苷的回收率較高[8]。
本方法中羥可酮的回收率很高,因為其最近已被FDA批準作為一種高效口服阿片類鎮痛劑[9],所以還有必要對羥可酮進行進一步研究。用本方法還能測定一些其它阿片類代謝物。曾有報道,氫可酮代謝為氫嗎啡酮,但也代謝為二氫可待因和norhydrocodone。羥可酮代謝為羥嗎啡酮,也代謝為a-和b-oxycodol、noroxycodol,以及其它產物。選擇哪種代謝物進行測定是比較復雜的。如前所述,高效阿片代謝物可能仍然具有其前體藥物的作用,但最近Danny Shen 博士實驗室有關羥可酮的數據對這一說法提出了質疑[10]。即使代謝物不具備其前體的藥學活性,它們也還可能有毒理學意義,例如,有助于區分急性和慢性使用藥物。
測定另一個可卡因代謝物芽子堿甲酯時,用本文報道的固相萃取提取,但回收率不穩定,原因可能是在蒸發過程中有損失。由于回收率容易改變,用本方法對芽子堿甲酯進行定量分析需要使用氘代內標。改進本方法的另一個辦法是使用shim 噴霧器(安捷倫部件號G1946-20307),這是為流動相流速高時提高離子傳輸進入毛細管的效率而設計的。可以提高本方法的分析靈敏度,使用的流動相流速為1.0 mL/min。
結論
本文介紹了一種分析血樣中阿片類化合物、可卡因和可卡因代謝物的方法,樣品經混合固定相固相萃取后,用電噴霧單級四極桿LC/MS 測定。本方法優于我們以前報道的GC/MS 方法,不需要衍生化,檢出限和定量限更低,所能測定的阿片類化合物范圍更寬。另外,在較高的溫度下使用高效ZORBAX Eclipse PlusC18 HPLC 柱,克服了以前遇到的阿片類化合物峰形差的問題。
