2018上半年FDA批準的17個新藥匯總及逐個評述
2018年上半年,FDA藥品審評與研究中心(CDER)批準了17個新藥,這些新藥包括新藥申請NDA中的新分子實體(NME)和生物制品許可申請(BLA)。

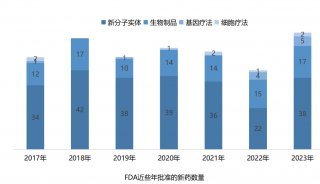
上圖是過去10年中,CDER每年批準的新藥數。2018年上半年CDER批準的新藥包括5個BLA和12個NME。相比于2017年上半年的23個減少了6個,略高于2016上半年和2015上半年。
罕見病/孤兒藥
(Rare OR "Orphan" Diseases)
已批準的17個新藥中,8個新藥獲得了孤兒藥資格(O),占CDER批準新藥的47%。罕見病有兩個特征,其一為患者人數少,美國為少于20萬人的疾病,歐盟屬于5/10000的疾病;其二為危及生命和健康的嚴重疾病。
優先審評(Priority Review)
如果CDER確定藥品能夠有潛力對醫療保健做出實質性推動,藥品將獲得優先審評。藥品在6個月內而不是標準的10個月內審評。2018年上半年獲批新藥中有8個被認定為優先審評(P),占17個新藥的47%。
CDER應用多種監管方法加快新藥研發和審批。這些方法除了優先審評還包括:快速通道(Fast Track)、突破性治療認定(Breakthrough)和加速批準(Accelerated Approval)。
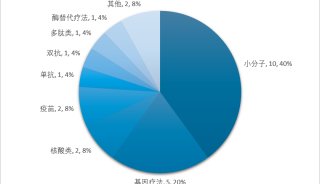
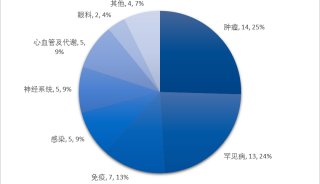
治療領域方面,2018年上半年還是比較豐富,抗腫瘤藥明顯減少,僅有2個。整體來說,美國FDA還是各藥企新藥上市的首選,17個新藥有12個藥物都是全球首批,其余的大多在歐盟首批。
下表為2018年批準的17個新藥匯總,來源于FDA網站,Novel Drug Approvals for2018。
品種評述
01 Lutathera
(lutetium Lu 177 dotatate)
1月26日,FDA批準了諾華子公司法國Advanced Accelerator Applications 公司Lutathera用于治療影響胰腺或胃腸道的一類癌癥,即胃腸胰腺神經內分泌腫瘤(GEP-NETs)。
Lutathera是一種Lu-177標記的生長抑素類似物,屬于新興的肽受體放射性核素療法(PRRT),通過與一種稱為生長激素抑制素受體的細胞結合而起作用,該生長抑素受體可能存在于某些腫瘤上。在與受體結合之后,藥物進入細胞,釋放輻射來損傷腫瘤細胞。
在美國和歐盟,Lutathera均被授予孤兒藥地位。歐盟已于2017年10月批準用于治療不可切除或轉移的生長抑素受體陽性胃腸胰腺神經內分泌腫瘤成人患者。這也是放射性藥物首次被FDA批準用于治療GEP-NETs。
02 Biktarvy
(bictegravir, embitcitabine,
tenofovir alafenamide)
2月7日,FDA批準了Gilead公司的Biktarvy,作為每日一次的單片片劑療法,用于治療HIV-1感染。
Biktarvy是一款全新的無助推(unboosted)整合酶鏈轉移抑制劑(INSTI),由bictegravir(50mg)、emtricitabine(200mg)、與tenofovir alafenamide(25mg)三種成分組成。
Biktarvy的療效與安全性在4項正在進行的3期臨床試驗中得到了驗證。試驗1489和1490招募的是初治的HIV-1感染成人患者,而試驗1844與1878則招募了病毒感染在病毒學上得到抑制的成人患者。
在試驗1489中,629名患者以1:1的比例分為兩組,分別接受Biktarvy與abacavir/dolutegravir/lamivudine(600/50/300mg)的治療。在48周后,這兩組患者中,分別有92.4%和93.0%的患者達到了HIV-1 RNA小于每毫升50c的主要終點。而在試驗1490中,645名患者接受的分別是Biktarvy與dolutegravir/FTC/TAF。同樣,兩組達到主要終點的比例接近,分別為89.4%與92.9%。
在試驗1878中,577名在藥物作用下,HIV-1 RNA已經小于每毫升50c的成人患者被1:1分為兩組,一組繼續現有的療法,另一組則切換到Biktarvy的治療。在48周后,兩組中均有1.7%的患者其HIV-1 RNA回到不低于每毫升50c的水平;而根據FDA的算法,分別有92.1%(Biktarvy組)與88.9%(現有療法組)的患者保持了HIV-1 RNA小于每毫升50c。
03 Symdeko
(tezacaftor; ivacafto)
2月13日,FDA批準了Vertex醫藥公司的Symdeko上市,用于治療12歲及以上的囊性纖維化(cystic fibrosis,CF)患者。SYMDEKO是Vertex獲得FDA批準的第3種針對囊性纖維化根本病因的治療藥物。
囊性纖維化是一種罕見的會縮短壽命的遺傳疾病,影響了北美、歐洲和澳大利亞約 75000 名患者,由基因突變導致的 CFTR 蛋白缺陷或缺失引起。兒童必須遺傳到兩個有缺陷的 CFTR 基因——分別來自父母,才會患囊性纖維化。CFTR 基因中有約 2000 個已知的突變,其中一些突變會通過在細胞表面產生不工作或過少的 CFTR 蛋白,它們可以通過基因檢測或基因分型檢測來確定。CFTR 蛋白的功能缺陷或缺失導致許多器官細胞中鹽和水的流入和流出不均衡。在肺中,這會造成異常粘稠的粘液積聚,引起慢性肺部感染和進行性肺損傷,并最終導致死亡。囊性纖維化患者的中位壽命在 20 歲左右。
據悉,歐洲藥品管理局(EMA)已經確認了tezacaftor/ivacaftor組合藥物的營銷授權申請(MAA)。公司預計在2018年下半年獲得歐盟批準。
04 Erleada(apalutamide)
2月14日,FDA批準強生Erleada (apalutamide) 上市,用于治療非轉移性(前列腺癌細胞未擴散)去勢抵抗(激素治療后疾病仍進展)的前列腺癌。
Erleada獲得過FDA的優先審評資格,是FDA批準的首個治療非轉移性去勢抵抗前列腺癌的藥物,也是首個憑借無轉移生存期(metastasis-free survival,MFS)的臨床終點獲批上市的腫瘤新藥。
Erleada的安全性及療效在涉及1207例非轉移性去勢抵抗前列腺癌的隨機研究中得到證實。患者隨機給予apalutamide或安慰劑,并接受內分泌治療,包括接受促性腺激素釋放激素(GnRH)類似物,或者手術去勢(切除雙側睪丸)以降低體內雄激素水平。結果顯示,apalutamide治療組無轉移生存期相比安慰劑組顯著延長(40.5 vs 16.2個月)。
05 Trogarzo(ibalizumab-uiyk)
2018年3月7日,美國FDA宣布正式批準藥明生物合作伙伴中裕新藥(TaiMed)的Trogarzo(ibalizumab-uiyk)上市,作為一種全新的抗逆轉錄病毒療法,治療現有多種療法均無法起效的成人HIV感染者。值得一提的是,這是美國FDA在2018年批準的首款創新生物藥,此項目也是藥明生物首個商業化生產的項目,標志著藥明生物躋身成為全球少數幾個通過FDA GMP認證的生物藥合作研發生產服務商(CDMO)。
Trogarzo是一種靜脈滴注的人源化免疫球蛋白G4單克隆抗體,它與CD4+ T細胞受體的第二個胞外區域結合,阻止HIV病毒入侵這些細胞。
Trogarzo的安全性與療效在一項臨床試驗中得到了驗證。該試驗招募了40名感染有多重耐藥性HIV的患者,他們均重度經治,有些患者甚至已經接受過10種或更多的抗逆轉錄病毒療法。然而即便接受了大量治療,他們血液中的病毒水平(HIV-RNA)依舊很高。研究人員發現,在現有的療法中額外加入Trogarzo的治療后,只要短短一周,大部分患者血液中的HIV-RNA水平就有顯著下降。24周后,43%的患者其HIV-RNA水平依舊得到了抑制。對于這些急缺治療方案的患者,Trogarzo帶來了顯著益處。
06 Ilumya(ibalizumab-uiyk)
3月20日,FDA批準ILUMYA (tildrakizumab-asmn)上市,用于適合接受全身治療或光療的中重度斑塊型銀屑病成人患者。
據3期臨床試驗數據顯示,與安慰劑相比,施用ILUMYA 100 mg能取得顯著的臨床改善。研究通過兩次用藥后第12周至少75%的皮膚清除率(銀屑病面積敏感指數或PASI 75),以及醫師全面評估(PGA)評分“清除”或“最小”進行測量。其中,該研究有74%(229人)的患者在三次用藥后第28周達到75%的皮膚清除率,84%持續用藥的患者在第64周能維持PASI 75。毋庸置疑,ILUMYA是中度至重度斑塊型銀屑病臨床治療上的又一個重要突破。
07 Tavalisse(fostamatinib)
4月17日,美國FDA批準了Rigel制藥的TAVALISSE用于對之前治療緩解不佳的成年慢性免疫性血小板減少癥(ITP)患者血小板減少的治療。TAVALISSE是一種口服的脾臟酪氨酸激酶(SYK)抑制劑,通過阻止血小板的破壞來應對疾病的潛在自身免疫原因,為成年慢性ITP患者提供了一個重要的新的治療方案。
TAVALISSE的批準依賴于FIT臨床研究項目的數據,FIT包含兩項隨機安慰劑對照的臨床3期研究047和048、一項開標擴展試驗049以及最初的概念驗證試驗。新藥上市申請包括了163例ITP患者的數據,并得到了一項安全數據集的支持,該數據集包含4600多名涉及其它適應癥的已接受TAVALISSE評估的受試者。
08 Crysvita(burosumab-twza)
4月17日,美國FDA批準了Ultragenyx Pharmaceutical公司的新藥Crysvita,成為首個獲批治療1歲及以上兒童和成年人的X連鎖低磷血癥(XLH)的藥物。XLH是一種罕見的遺傳性軟骨病,會造成血液中磷含量低,導致兒童和青少年的骨骼生長和發育受損,并使其一生都有骨礦化的問題。
Crysvita獲批是基于4項臨床試驗中的安全性和療效數據。在一項安慰劑對照臨床試驗中,94%的每月接受一次Crysvita的成年人能達到正常的磷水平,而安慰劑組患者只有8%能達到這一水平。在兒童中,每兩周接受一次Crysvita的患者中有94%到100%能達到正常的磷水平。在兒童和成人中,接受Crysvita療法的患者的X光(與XLH相關)結果有所改善。把這些結果與自然歷史隊列相比較,也為Crysvita的有效性提供了支持。
09 Akynzeo (fosnetupitant
and palonosetron)
4月19日,FDA批準了瑞士制藥集團Helsinn公司的靜脈注射AKYNZEO,作為經歷CINV的患者的替代治療選擇。
FDA已批準將AKYNZEO?IV與成人地塞米松聯合用于預防與高度致吐性癌癥化療的初始和重復過程有關的急性和延遲性惡心和嘔吐。尚未研究AKYNZEO?用于預防與蒽環類加環磷酰胺化療相關的惡心和嘔吐
口服AKYNZEO在2014年被美國食品藥物管理局批準為固定組合口服藥,用于預防與癌癥化療初期和重復過程有關的急性和延遲性惡心和嘔吐,包括但不限于高度致吐化療。
AKYNZEO?靜脈注射制劑的批準為將這種重要治療選擇帶入更多患者采用新配方鋪平了道路,并將于2018年5月在美國推出此產品。
10 Lucemyra
(ofexidine hydrochloride)
5月16日,美國FDA宣布批準US WorldMeds的Lucemyra(鹽酸洛非斯汀,lofexidine hydrochloride),用于緩解突然停用阿片類藥物的成人患者的戒斷癥狀。雖然Lucemyra可以減輕戒斷癥狀的嚴重程度,但它無法完全阻止這些癥狀,并且最長只能使用14天。
Lucemyra的安全性和有效性得到了兩項隨機、雙盲、安慰劑對照臨床試驗的支持。這些臨床試驗共有866名符合阿片類藥物依賴診斷標準的成人患者,這些患者在生理上依賴于阿片類藥物,并正在進行阿片類藥物戒斷。這些研究使用Gossop短暫停藥量表(SOWS-Gossop)來評估療效,這是一種通過患者報告結果來評估阿片類戒斷癥狀的工具。這些癥狀包括感覺不適、胃痙攣、抽搐、感覺冷、心臟劇烈跳動、肌肉緊張、疼痛,打呵欠、流淚和失眠。
11 Aimovig(erenumab-aooe)
5月17日,FDA批準了安進(Amgen)公司的Aimovig,作為成人偏頭痛的預防性治療,給藥方式為每月一次的自我注射,這也是FDA批準的首個預防性偏頭痛治療藥物。
Aimovig獲批主要基于圍繞其展開的三項臨床試驗,試驗評估了其在偏頭痛預防中的安全性和有效性。
第一項研究包括955名有發作性偏頭痛病史的參與者,并將Aimovig與安慰劑進行比較。在6個月的時間里,aimovig治療的患者平均每月的偏頭痛天數比服用安慰劑的患者少1到2個月。
第二項研究包括577名有發作性偏頭痛病史的患者,并將Aimovig與安慰劑進行比較。在三個月的時間里,aimovig治療的患者平均每個月比服用安慰劑的病人少一次偏頭痛。
第三項研究對667例慢性偏頭痛患者進行了評估,并將Aimovig與安慰劑進行比較。在這項研究中,在三個月的時間里,接受Aimovig治療的患者平均每月比接受安慰劑的患者少2次。臨床試驗中最常見的副作用是注射部位的反應和便秘。
12 Lokelma (sodium zirconium cyclosilicate)
5月18日,美國FDA批準了阿斯利康(AstraZeneca)的新藥Lokelma用于治療罹患高鉀血癥(hyperkalaemia)的成人患者。
高血鉀風險在慢性腎病(CKD)患者以及服用普通心力衰竭(HF)藥物(如腎素-血管緊張素-醛固酮系統[RAAS]抑制劑)的患者中顯著增加,因為這種藥物會增加血鉀水平。
Lokelma于今年3月底在歐盟獲批上市。次Lokelma獲得FDA的批準是基于三項雙盲、安慰劑對照試驗和兩項開放標簽試驗數據的支持。
這些研究表明,Lokelma的起效時間是服藥后1.0小時,達到正常血鉀水平的中位時間是2.2小時,92%的患者在基線后48小時內達到正常血鉀水平。該藥物的治療效果可維持長達12個月。
13 Doptelet(avatrombopag)
5月21日,美國FDA批準Dova Pharmaceuticals子公司AkaRx的新藥Doptelet片劑,用于治療計劃接受醫療或牙科手術的慢性肝病(CLD)成人患者的低血小板計數(血小板減少癥)。
Doptelet的安全性和有效性在兩項試驗(ADAPT-1和ADAPT-2)中得到了驗證。這些研究共包含435名慢性肝病和嚴重血小板減少癥患者,他們將接受通常需要輸注血小板的手術。這些試驗評估了兩個劑量水平的口服Doptelet與安慰劑相比治療5天的效果。結果顯示,與安慰劑組相比,兩種劑量水平的Doptelet組有較高比例的患者具有增加的血小板計數,并且不需要在手術當天和治療后7天內接受血小板輸注或任何救援治療。Doptelet最常見的副作用有發燒、胃(腹)痛、惡心、頭痛、疲勞和手足腫脹(水腫)。
14 Doptelet(avatrombopag)
5月24日,FDA批準了BioMarin Pharmaceutical公司的Palynziq注射劑,用于降低苯丙酮尿癥(PKU)成人患者的血液苯丙氨酸(Phe)水平 ,這些患者在現有管理下其血液Phe濃度無法控制在600微摩爾/升以內。
Palynziq是一種聚乙二醇化的重組苯丙氨酸解氨酶,用來替代PKU患者缺乏的苯丙氨酸羥化酶(PAH)以分解Phe。在關鍵3期研究PRISM-2中,Palynziq與安慰劑相比顯著降低了血液Phe水平(p<0.0001),抵達了血液Phe變化的主要終點。在PRISM-2雙盲、安慰劑對照、隨機停藥期試驗(RWP)中,患者以2:1的比例被隨機分配繼續接受Palynziq治療(每日20 mg或每日40 mg)或接受安慰劑,持續8周。結果顯示,Palynziq組患者可以維持血液Phe濃度,而安慰劑組患者的血液Phe濃度恢復到治療前基線。
15 Olumiant(baricitinib)
5月31日,美國FDA批準了禮來公司(Eli Lilly)新藥Olumiant上市,治療罹患中度至重度類風濕關節炎,卻無法從TNF抑制劑治療中受益的成人患者。
由禮來與Incyte帶來的Olumiant就是這樣一款充滿潛力的新藥。它是一款每日一次的口服JAK抑制劑,能高效抑制JAK1、JAK2、以及TYK2。在人體內,不少細胞因子依賴于JAK的活性,其在不少炎性疾病和自身免疫疾病的發病過程中有潛在作用。通過抑制多種JAK的活性,Olumiant有望給類風濕關節炎患者帶來福音。
16 Epidioloex (cannabidiol)
6月25日,美國FDA批準了GW RESEARCH LTD新藥Epidiolex口服溶液治療兩歲及以上患者的兩種罕見和嚴重癲癇,Lennox-Gastaut綜合征和Dravet綜合征相關的癲癇發作。這是FDA批準的第一種含有從大麻中提取的純化藥物的藥物。這也是FDA首次批準用于治療Dravet綜合征患者的藥物。
在三項隨機,雙盲,安慰劑對照的臨床試驗中研究了Epidiolex的有效性,該試驗涉及516名患有Lennox-Gastaut綜合征或Dravet綜合征的患者。與安慰劑相比,Epidiolex與其他藥物一起被證明可有效降低癲癇發作的頻率。
17 Zemdri (plazomicin)
6月25日,美國FDA批準了Achaogen新藥Zemdri上市,用于由某些腸桿菌科細菌感染引起的、治療選擇非常有限或無治療選擇的復雜性尿路感染(cUTI,包括腎盂腎炎)成人患者,該藥是一種靜脈輸注藥物,每天給藥一次。
此次批準,使Zemdri成為治療cUTI的唯一一種每日一次的氨基糖苷類療法。plazomicin是一種新一代的氨基糖苷類抗生素,能夠抑制細菌蛋白質的翻譯過程。plazomicin是在西梭霉素(sisomicin)基礎上進行了化學改造而得,能避免被主要的氨基糖苷類抗生素鈍化酶(AME)破壞而失去活性。plazomicin開發用于治療MDR革蘭氏陰性菌腸桿菌科細菌導致的嚴重感染,包括對碳青霉烯類抗生素耐藥的腸桿菌。
Zemdri的獲批,是基于III期臨床研究EPIC的數據。該研究是首個評估每日一次氨基糖苷類療法治療cUTI(包括腎盂腎炎)的隨機對照研究,數據顯示,Zemdri達到了與美羅培蘭(meropenem)的非劣效性。