空間蛋白質組學:一種強大的細胞生物學發現工具
真核細胞高度區室化,生物過程被分隔在不同的區室進行。蛋白質功能與亞細胞定位密切相關,不同的區室提供不同的化學環境(例如pH和氧化還原條件)、不同的潛在作用配體或底物。因此,對蛋白質亞細胞定位的嚴格控制是細胞生理學的重要調控內容。
大多數細胞生物學過程涉及蛋白質亞細胞定位的變化,例如轉錄因子在細胞核-胞漿的穿梭、細胞凋亡過程中線粒體蛋白的重新定位,以及細胞表面信號傳導受體的內吞等。相反,蛋白質的錯誤定位通常與細胞功能障礙和疾病相關,包括神經變性、癌癥和代謝紊亂等。
以蛋白質空間定位為研究方向的空間蛋白質組現在已經用于揭示人類蛋白質組的復雜結構,如單細胞變異、動態蛋白質易位,相互作用網絡改變,以及蛋白定位改變等。一些研究者也已成功運用空間蛋白質組學來研究疾病,包括急性病毒感染、肝病等。
2019年5月,KTH 瑞典皇家理工學院的 Emma Lundberg 教授和德國馬克斯-普朗克研究所的 Georg H. H. Borner 教授,在國際著名期刊 Nature Reviews | Molecular Cell Biology(IF=35.612)發表了題為《Spatial proteomics: a powerful discovery tool for cell biology》的綜述性文章,系統介紹了空間蛋白質組學的技術、未來發展的機遇與挑戰。下面小編為大家解讀一下這篇綜述。

目前三種互補的方法可用于空間蛋白質組學研究:細胞器分級的質譜分析(圖1)、蛋白質與蛋白質互作網絡分析(圖2),以及基于蛋白質定位的蛋白質成像(圖3)。
1)基于質譜的細胞器分級方法
質譜可用于復雜混合物中蛋白質的定性和定量研究。空間蛋白質組學可借助傳統生物化學分析方法,如下圖1a 所示,通過定制的亞細胞分級分離(如,梯度離心或差速離心)來富集目標細胞器(綠色)。然后利用質譜方法只分析富集到的組分。此方法鑒定出來的蛋白質包含目標細胞器蛋白和共富集污染物(紅色和藍色),且無法區分,因而不推薦。
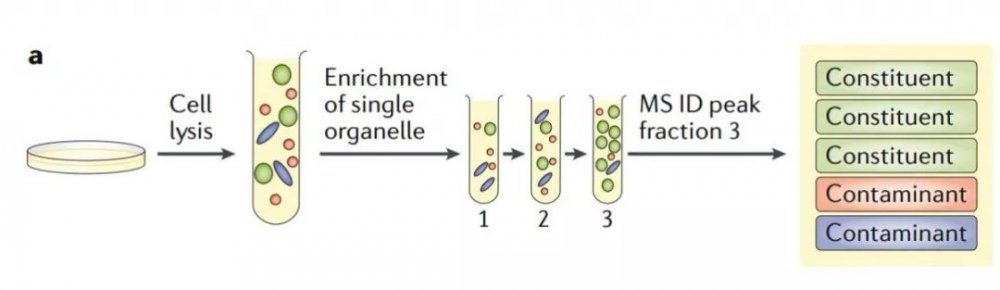
圖1a 空間蛋白質組學基于質譜進行細胞器分級分析
下圖1b 為單細胞器分析法,定量質譜方法可對目標餾分和某幾個相鄰或相近餾分進行檢測。對于每種蛋白質都可獲得豐度分布曲線。此方法可通過統計分析區分污染物(紅色和藍色),污染物可被識別,但卻不一定能分解為不同的類別。
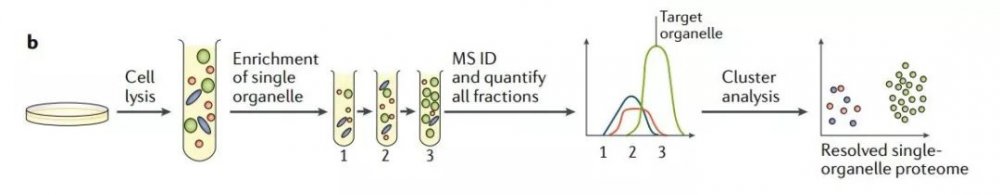
圖1b 單細胞器分析法
下圖1c 為多細胞器分析法,應用亞細胞分級方案,同時分離所有的細胞器。由于沒有進行細胞器“純化”,因而細胞器很大程度上重疊分布。之后對亞細胞分級進行定量質譜分析。每個細胞器都有自己獨特的蛋白輪廓,通過對蛋白聚類分析,并使用已有的細胞器標記,對聚類蛋白進行注釋。
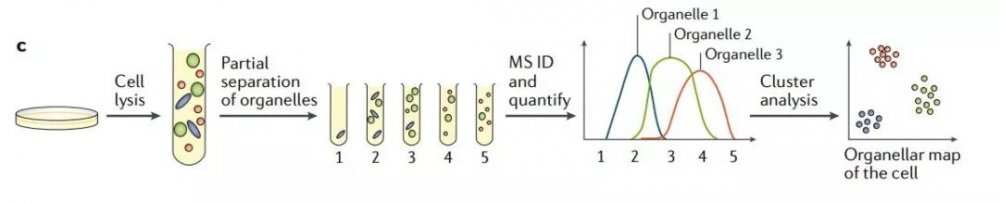
圖1c 多細胞器分析法
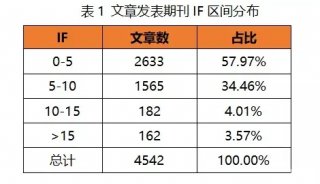
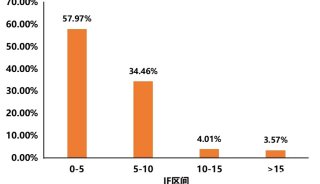
目前基于多細胞器分析的質譜方法,在細胞器分離方法、質譜定量策略,以及生物信息分析方法上,衍生出多種技術改進方案。不論何種方案,過去3年發表的多細胞器分析的應用中,都實現了非常高水平的細胞器分辨率(≥10個亞細胞區室)、蛋白質組學覆蓋率(> 5,000個蛋白質),以及分類的準確率(通常> 90%)(表1)。
表1.基于質譜的多細胞器分析的當前實施方式

另外值得一提的是,由于質譜的高分辨率,細胞器質譜圖本質上提供了大量潛在的相互作用的組學數據。如,作為同一復合物的蛋白質在細胞器圖譜上顯示為微團簇,這一特征可用于鑒定新型蛋白質復合物(圖1d)。此外,細胞器圖譜原則上可提供肽段水平的分辨率(圖1e),并揭示不同的蛋白質剪接體、蛋白水解加工形式,以及與翻譯后修飾相關的多種定位差異。隨著質譜技術的進一步發展,基于肽水平的細胞器定位可能會為解開蛋白質亞細胞定位的復雜性做出重大貢獻。
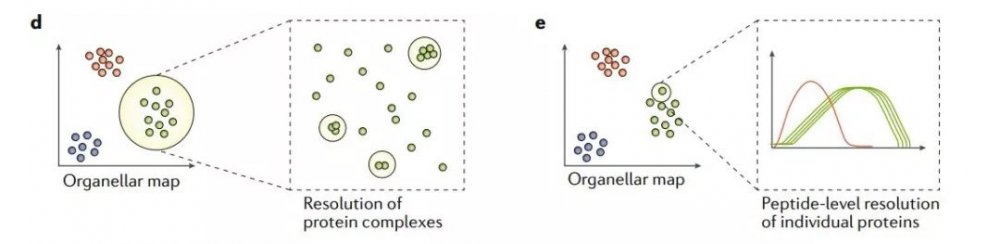
圖1de 空間蛋白質組學基于細胞器質譜圖進行細胞器分級分析
2)基于蛋白互作網絡的空間蛋白質組研究方法
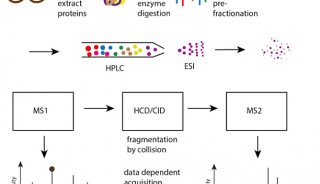
蛋白質-蛋白質相互作用普遍使用基于質譜的、抗體介導的親和純化-MS(AP-MS)來進行研究。使用抗體從復雜樣本(如全細胞裂解物)中親和純化蛋白質及其結合配體,后續 MS 分析鑒定(圖2a)。
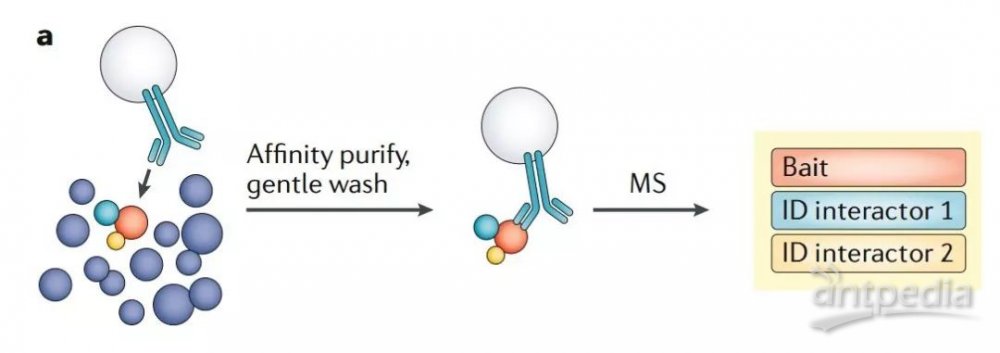
圖2a 通過蛋白互作網絡研究空間蛋白質組學
從概念上將,由于相互作用的蛋白必須位于相同的亞細胞位置,所以蛋白質的相互作用組可以認為是“局部”的空間蛋白質組。使用相互作用的誘餌蛋白進行多個 AP-MS 實驗,也可以揭示一個具有空間信息的蛋白關聯網絡(圖2b)。
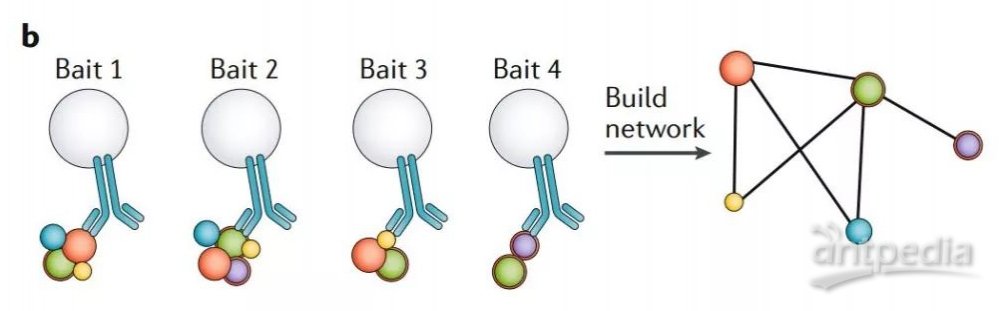
圖2b
鄰近標記法對于細胞器的 AP-MS 十分有幫助。誘餌蛋白可以使用人工合成的抗壞血酸過氧化物酶(APEX)或生物素連接酶(BioID)進行標記。生物素連接酶可以催化誘餌蛋白緊鄰(<10-20nm)蛋白質的生物素化。因而質譜分析的靶標蛋白不僅包括誘餌蛋白、與誘餌蛋白直接結合的配體,還包括瞬時相互作用的蛋白,以及位置緊密相連但不直接結合的蛋白(圖2c)。由于少量誘餌蛋白便可獲取豐富的空間信息,因而此方法可以在不需要亞細胞分級的情況下獲得全面的區室蛋白質組(圖2d)。
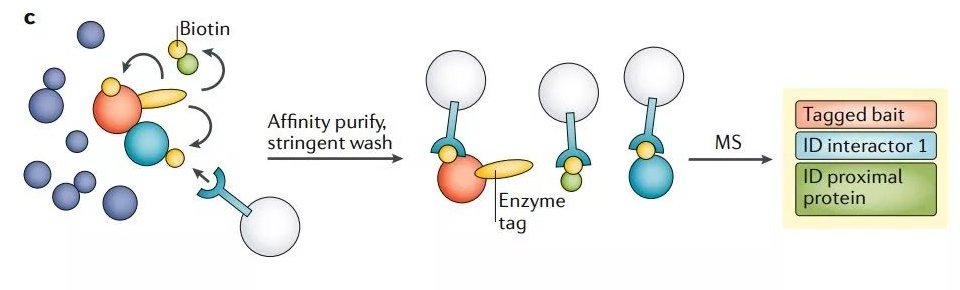

圖2cd?
當在同一系統中進行多個 AP-MS 或鄰近標簽標記實驗時,誘餌蛋白和結合配體會在不同實驗中重復出現(如在b部分中)。來自相同亞細胞定位的多個誘餌蛋白會揭示各個區室的詳細圖譜,而區室與區室又通過相互間的映射連接,最終便可以構建細胞全部亞細胞區室的空間蛋白質圖譜(圖2e)。

圖2e?
3)基于成像的空間蛋白質組方法
基于成像的空間蛋白質組學提供了在自然細胞環境下可視化研究蛋白質的機會,而不需要在蛋白質組學分析之前進行細胞裂解或細胞器的分離(圖3)。基于成像的蛋白質定位便于研究具有多模式細胞器定位的蛋白質研究,實際上大多數的蛋白質也是定位于多個亞細胞區室的。此外,越來越多的研究表明,遺傳背景相同的細胞群體也會在蛋白表達水平、蛋白定位上表現為差異。基于成像的空間蛋白質組學技術有助于通過捕獲單細胞分辨率下的蛋白空間分布來研究這種可變性。蛋白質的可視化成像,通常使用抗體,或熒光蛋白的融合表達來實現。
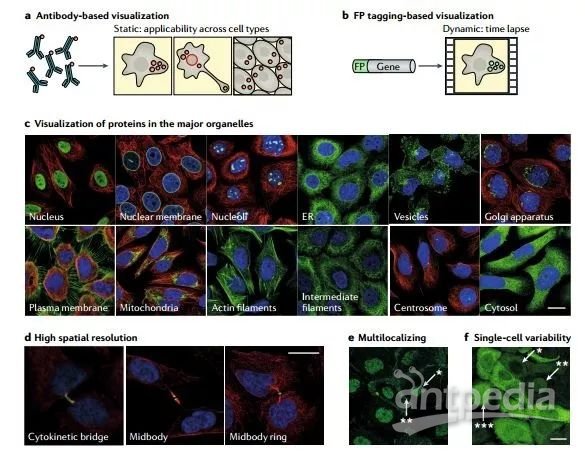
圖3. 基于成像的空間蛋白質組的多種處理方式
許多因素使得細胞蛋白質組比僅從基因數目進行的蛋白預測,要復雜的多。空間蛋白質組則最有希望解開這種有趣的細胞復雜性。
1)幫助識別多定位和“兼職”蛋白,了解細胞復雜功能
約50%的人類蛋白是定位于多個亞細胞區室的。這些蛋白質是否具有特定的兼職功能?“兼職”(Moonlighting)蛋白被定義為具有兩種或更多不同細胞功能的蛋白質,這些細胞功能不是由遺傳變異、RNA 剪接或基因多重效應引起的,而是由亞細胞定位,底物,寡聚化或翻譯后修飾(PTM)的差異引起的。已知的兼職蛋白數量正在迅速增加。據估計,23%的人體蛋白質是兼職蛋白。78%的已知兼職蛋白參與疾病發生發展,48%是目前的藥物靶點。基于成像的空間蛋白質組學將是識別多定位蛋白質的關鍵技術,以更好地理解它們在復雜細胞功能中的關鍵作用。
2)幫助發現多種蛋白質存在形式(Proteoforms)
蛋白質的每種分子形式都稱之為一種蛋白質形式,這種變化主要是由于 DNA 序列的變異性,RNA 剪接和不同的翻譯后修飾,如磷酸化,泛素化,烷基化和糖基化所引起的。典型的人類細胞預估含有600萬個共存的蛋白質形式。雖然這個數字遠遠低于理論上可能的組合數量,但它揭示了蛋白質組的巨大復雜性。基于質譜(MS)的空間蛋白質組學,可以提供關于蛋白質存在形式和亞細胞定位的新見解。
3)獲取蛋白質豐度信息
人體細胞蛋白豐度跨越7個數量級,為了模擬細胞的生物學過程,需要了解每個細胞的絕對蛋白的數目及其變化。基于質譜和基于成像的空間蛋白質組學,可以提供這些信息。
4)獲得蛋白質層面的單細胞變異性信息
越來越清楚的是,遺傳上相同的細胞群顯示出蛋白質表達的變異。我們對這種細胞異質性的后果的理解仍然不成熟。基于成像的方法可能在闡明致病因素方面起關鍵作用。
5)了解蛋白質定位的動態信息
蛋白質亞細胞定位受到嚴格控制,許多蛋白質響應刺激,擾動或疾病而改變定位。全球比較空間蛋白質組學,基于成像,基于相互作用或基于 MS,提供了在系統水平捕獲這些生理和病理蛋白質易位的理想工具,并且應該成為細胞生物學家的廣泛發現工具。
綜上,基于質譜與成像的空間蛋白質組有助于全面了解細胞的復雜性。而且后續空間蛋白質組學與其他組學的結合,也將利于發現更多的生物信息。例如,轉錄組學可以提供細胞特異性表達,如剪接變體、單核苷酸多態性等數據信息,從而使空間蛋白質組學能夠發現新的蛋白質形式。空間蛋白質組學與代謝組學的結合,可以在功能上發現細胞器重排與代謝變化的相關性。與之類似地,RNA 測序與空間蛋白質組學相結合,有可能將 mRNA 與亞細胞定位聯系起來。因此作者認為,空間蛋白質組學應該成為細胞繪圖工作中的整合技術,例如人類細胞圖譜,旨在表征所有人類細胞類型。
參考文獻:
Spatial proteomics: a powerful discovery tool for cell biology.?Lundberg E,?Borner GHH. Nat Rev Mol?Cell?Biol.?2019 May;20(5):285-302. doi: 10.1038/s41580-018-0094-y.
-
焦點事件

-
企業風采

-
精英視角

-
項目成果
-
綜述
-
焦點事件